Introduction
Cholangiocarcinoma, also known as bile duct cancer,
is a highly aggressive and metastatic cancer derived from
epithelial cells lining the intrahepatic or extrahepatic bile ducts
(1). Due to the presence of
metastasis at diagnosis for the majority of patients, rendering the
tumour inoperable, the overall prognosis for cholangiocarcinoma is
poor, and the five-year survival is 5-10% (2). Intensive efforts have been dedicated
to developing effective therapies, particularly those targeting the
metastatic spread of cholangiocarcinoma.
Cancer metastasis is a multi-step process that
involves the local migration and invasion of tumour cells followed
by distant dissemination of these cells through blood and lymphatic
vessels. Metastatic phenotypes gained by tumour cells, coupled with
enhanced angiogenesis, predispose malignant cancer development
(3). Epithelial-mesenchymal
transition (EMT) is a biological process through which epithelial
cells acquire morphological, structural, and functional features of
mesenchymal cells, characterized by the downregulation of cell-cell
junction components, including E-cadherin and β-catenin, the
upregulation of mesenchymal markers, including N-cadherin and
catenin, and enhanced migratory and invasive capacities (4,5). EMT
has been well demonstrated to be essential for the malignant growth
and the metastatic spread of multiple cancer types, including
cholangiocarcinoma (6,7). Therefore, characterizing and
targeting the molecular mechanisms underlying EMT and angiogenesis
may be beneficial for developing treatment strategies against
cholangiocarcinoma.
Leptin is an adipocyte-derived cytokine (adipokine)
that signals through specific leptin receptors, activates various
downstream signalling pathways, and serves multiple roles in human
physiology and pathology (8). The
well-documented association between obesity and cancer has directed
attention towards leptin. Studies over the past decade have
reported a positive association between serum leptin levels and
multiple cancer types, including breast (9), lung (10), pancreatic (11), and gastrointestinal cancer
(12). Furthermore, the signalling
mechanisms and functional significance of leptin during the
development of multiple cancer types have been demonstrated
(13). The primary signal
transduction pathways downstream of leptin receptors include Janus
kinase 2 (JAK2), phosphoinositide 3-kinase (PI3K), SH2-containing
protein tyrosine phosphatase 2/mitogen-activated protein kinase
(SHP2/MAPK), 5′-AMP-activated protein kinase/acetyl-CoA carboxylase
(AMPK/ACC), and mammalian target of rapamycin (mTOR) kinase
(14). A recent study on multiple
breast cancer cell lines demonstrated that leptin, through the
activation of the PI3K/AKT signalling pathway, upregulates pyruvate
kinase muscle isozyme M2 (PKM2), a pyruvate kinase isoform specific
to embryonic development and absent in the majority of adult
tissues (15). PKM2 is
overexpressed in breast cancer cells and essential for
leptin-induced EMT in these cells (15). Similarly, PKM2 is upregulated in
other cancer types, and functionally important for multiple
phenotypes, including cancer metabolism, tumour growth and EMT
(16,17). In addition to acting on cancer
cells, leptin increases the expression of vascular endothelial
growth factor (VEGF) and promotes angiogenesis of endothelial cells
(18). Therefore, leptin is an
attractive target for cancer therapy.
Despite the abundant evidence regarding the
association between leptin and numerous cancer types, few studies
have assessed its significance in cholangiocarcinoma. Fava et
al (19) reported that, by
activating PI3K/JAK signalling, leptin stimulates the growth and
migration of cholangiocarcinoma cells. However, whether leptin is
essential for the malignant phenotypes of cholangiocarcinoma and,
if so, which mechanisms are responsible remain unclear. To address
these questions, the present study focused on EMT and angiogenesis,
assessed the significance of leptin in regulating these biological
behaviours, and explored the underlying molecular mechanisms.
Materials and methods
Cell culture and treatments
The human cholangiocarcinoma cell lines SK-ChA-1 and
TFK-1 and the human umbilical vein endothelial cells (HUVECs) were
purchased from Shanghai Institute of Biochemistry and Cell Biology,
Chinese Academy of Medical Sciences. The SK-ChA-1 cells were
cultured in Dulbecco's modified Eagle medium (DMEM) and TFK-1 cells
were cultured in RPMI-1640 (both from Gibco; Thermo Fisher
Scientific, Inc.), supplemented with 10% foetal bovine serum (FBS;
HyClone; GE Healthcare Life Sciences), 100 U/ml penicillin and 100
mg/ml streptomycin. HUVECs were cultured in endothelial cell growth
medium (PromoCell GmbH). All cell cultures were maintained at 37°C
in a humidified atmosphere with 5% CO2.
For leptin treatment, cells in the log phase were
treated with leptin (R&D Systems, Inc.) at 0, 50, 100 or 200
ng/ml, for 0, 24 or 48 h. The morphology of cells was imaged under
a light microscope (magnification, ×400).
Synthesis and transfection of RNA
oligoribonucleotides
Negative control siRNA (si-NC;
5′-GGCCAGACTGGGAAGAAAA-3′), siRNA targeting PKM2 (si-PKM2;
5′-CUUGUCCGAUGUUACCCAATT-3′), miRNA mimics negative control
(miR-NC; sense 5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense
5′-ACGUGACACGUUCGGAGAATT-3′), miR-122 mimics (sense
5′-UGGAGUGUGACAAUGGUGUUUG-3′ and antisense
5′-AACACCAUUGUCACACUCCAUU-3′), miRNA inhibitor negative control
(inhibitor-NC; 5′-CAGUACUUUUGUGUAGUACAA-3′) and inhibitor miR-122
(5′-CAAACACCAUUGUCACACUCCA-3′) were designed and synthesized by
Shanghai GenePharma Co., Ltd.
Transfection of siRNA, miRNA mimics or inhibitor was
performed using Lipofectamine 2000 reagents (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. In
brief, SK-ChA-1 and TFK-1 cells were seeded on 6-well plates
(5×105 cells/well) until ~75% confluency was achieved.
si-PKM2 (100 nM), miR-122 mimics (50 nM), miR-122 inhibitor (100
nM) or the corresponding scrambled sequence (100 nM; si-NC, miR-NC
and inhibitor NC, respectively) that served as negative control
were transfected into the cells in serum-free medium using
Lipofectamine 2000. At 48 h post-transfection, the levels of PKM2
and miR-122 were quantified using reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) to
evaluate the effect of transfection.
MTT assay
To assess the effects of leptin on cytotoxicity and
cell proliferation, TFK-1 cells were seeded into 96-well plates
(Corning Incorporated) in triplicate at 5×105 cells/100
μl/well. Upon treating the cells with leptin at 0, 100 or
200 ng/ml for 24 h or 48 h, 20 μl of MTT reagent (5 mg/ml)
was added to each well and incubated at 37°C for a further 4 h.
Following centrifugation at 250 × g for 5 min at room temperature,
the supernatant was discarded from each well. DMSO (150
μl/well) was added to each well to dissolve the formazan
crystals. The absorbance was measured using a microplate reader at
490 nm.
Transwell migration and invasion
assays
Transwell chambers coated with Matrigel matrix (BD
Biosciences) and uncoated Transwell chambers (8-μm pore;
Corning Incorporated) were used for the invasion and migration
assays, respectively. Briefly, 1×105 SK-ChA-1 or TFK-1
cells were seeded into the top chamber, and 600 μl of medium
containing leptin was added to the lower chamber. After 24 h, cells
that did not invade through the membrane were removed using a
cotton swab. Cells that invaded the membrane and attached to the
bottom side of the inserted membrane were stained with 1% crystal
violet at room temperature for 10 min, imaged and counted under a
light microscope (magnification, ×100).
Extraction of total RNA and RT-qPCR
Total RNA was extracted from SK-ChA-1 or TFK-1 cells
using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. cDNA was then synthesized
with M-MLV reverse transcriptase kit (Invitrogen; Thermo Fisher
Scientific, Inc.) according to manufacturer's protocol. Briefly, 1
μg total RNA and 1 μl dNTP mix (10 mM each dATP,
dGTP, dCTP and dTTP) were mixed and incubated at 65°C for 5 min.
Then, the reaction mixtures, including 4 μl 5X first-strand
buffer, 2 μl 0.1 mol DTT and 40 U of ribonuclease inhibitor
were incubated at 37°C for 2 min, followed by incubation with 1
μl M-MLV RT at 37°C for 2 min, 50 min at 37°C and heat
inactivation at 70°C for 15 min. qPCR was performed using
SuperScript® III Platinum® SYBR Green One-Step qRT-PCR kit
(Invitrogen; Thermo Fisher Scientific, Inc.) on the ABI PRISM 7300
Fast Real-Time PCR system (Ambion; Thermo Fisher Scientific, Inc.)
according to manufacturer's protocol. The thermocycling conditions
were as follows: Initial 3 min denaturation step at 95°C; followed
by 40 cycles at 95°C 3 sec for denaturation; and at 60°C for 30 sec
for annealing and extension. Primer sequences are listed in
Table I. Each reaction was
performed in triplicate, and the relative expression levels of each
target gene were calculated as a ratio to that of GAPDH, the
internal control, using the 2−∆∆Cq
method (20).
 | Table IPrimer sequences used for reverse
transcription-quantitative PCR. |
Table I
Primer sequences used for reverse
transcription-quantitative PCR.
| Gene | Sequence
(5′-3′) |
|---|
| hPKM2 | |
| Forward |
GAGTACCATGCGGAGACCAT |
| Reverse |
GCGTTATCCAGCGTGATTTT |
| hsa-miR-122 | |
| Forward |
AGCGTGGAGTGTGACAATGG |
| Reverse |
CGGCCCAGTGTTCAGACTAC |
| hβ-catenin | |
| Forward |
GAACTCTCGAAGGACCCAGC |
| Reverse |
TAAATCCCAAGAGGCCCTGC |
| hE-cadherin | |
| Forward |
TCACATCCTACACTGCCCAG |
| Reverse |
AGTGTCCCTGTTCCAGTAGC |
| hAng1 | |
| Forward |
GGGGGAGGTTGGACTGTAAT |
| Reverse |
GAATAGGCTCGGTTCCCTTC |
| hVEGFA | |
| Forward |
CACCAAGGCCAGCACATAGG |
| Reverse |
AGGGAGGCTCCAGGGCATTA |
| hN-cadherin | |
| Forward |
AGGGGACCTTTTCCTCAAGA |
| Reverse |
TCAAATGAAACCGGGCTATC |
| hVimentin | |
| Forward |
GGACCAGCTAACCAACGACA |
| Reverse |
AAGGTCAAGACGTGCCAGAG |
| GAPDH | |
| Forward |
GCTGTAGCCAAATCGTTGT |
| Reverse |
CCAGGTGGTCTCCTCTGA |
Western blot analysis
SK-ChA-1 or TFK-1 cells were collected and lysed
using cell lysis buffer (Beyotime Institute of Biotechnology).
Protein concentration was determined using a Pierce BCA Protein
Assay kit (Thermo Fisher Scientific, Inc.). Subsequently, 30
μg total protein/lane were boiled at 95-100°C for 5 min and
separated on a 12% SDS-PAGE gel, and then proteins were transferred
onto a polyvinylidene difluoride membrane. Upon blocking the
membrane in TBST buffer (10 mM Tris, pH 8.0, 150 mM NaCl, 0.5%
Tween-20) containing 5% non-fat milk at room temperature for 1 h,
the target protein was probed with one of the following primary
antibodies at 4°C overnight: Rabbit anti-human E-cadherin,
(1:1,000; cat. no. A3044), rabbit anti-human β-catenin (1:1,000;
cat. no. A10834), rabbit anti-human N-cadherin (1:1,000; cat. no.
A0433), rabbit anti-human vimentin (1:1,000; cat. no. A11952),
rabbit anti-human VEGFA (1:1,000; cat. no. A0280), rabbit
anti-human angiotensin 1 (Ang1; 1:1,000; cat. no. A945; all from
ABclonal Biotech Co., Ltd.), anti-PKM2 (1:1,000; cat. no. 4053;
Cell Signaling Technology, Inc.), or rabbit anti-human GAPDH
(internal control; 1:5,000, AC001, ABclonal Biotech Co., Ltd.). The
membrane was then incubated with horseradish peroxidase-conjugated
anti-rabbit IgG (1:5,000; cat. no. 7074; Cell Signaling Technology,
Inc.) at room temperature for 2 h. The signal was developed using
the Pierce™ Enhanced Chemiluminescence system (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. The
signal density was analysed using ImageJ software (version 1.51q;
National Institutes of Health), and the relative protein level was
calculated as the density ratio of the target protein to GAPDH
(internal control).
Tube formation assay
To assess in vitro angiogenesis, conditioned
medium was collected from cholangiocarcinoma cells. Briefly,
2×105 cells were seeded into 10-cm tissue culture
plates. Following an overnight culture, cells were treated with 0
or 200 ng/ml leptin for 24 h. Following three washes with DMEM,
cells were cultured in serum-free DMEM for a further 24 h. The
conditioned medium was collected and centrifuged at 2,000 × g for
10 min at 4°C to remove any cell debris. A 24-well plate was coated
with Matrigel. Upon gel solidification, HUVECs (1×105
cells/well) were seeded on top of the Matrigel in triplicate,
treated with a mixture of conditioned medium:EGM2 medium (volume
ratio 2:1), and incubated at 37°C with 5% CO2 for 6 h.
Each well was imaged under an Olympus DP71 microscope (Olympus
Corporation) at ×100 magnification, and the quantification of
branching points was performed using ImageJ software (version
1.51q).
Luciferase reporter assay
starBase (http://starbase.sysu.edu.cn/) and miRDB (http://mirdb.org) software tools were used to identify
potential miRNAs that may bind to the 3′-untranslated region
(3′-UTR) of the human PKM2 gene (21,22).
For the luciferase reporter assay, the 3′-UTR sequence of the PKM2
gene containing the potential miR-122-binding site was cloned into
a psiCHECK-2 luciferase reporter plasmid (Promega Corporation) and
transfected into TFK-1 cells using Lipofectamine 2000 according to
the manufacturer's protocol. Upon treating the cells with vehicle
control (PBS only), or transfecting miR-122 mimics or miR-122
inhibitor for 36 h, luciferase activity was detected using the Dual
Luciferase Reporter assay system (Promega Corporation) according to
the manufacturer's protocol. Renilla luciferase activity was
normalized to firefly luciferase activity in cells.
Collection of blood samples and
ELISA
The present study was approved by the Institutional
Review Board of Hunan Provincial People's Hospital, and written
informed consent was obtained from all participants. A total of 15
patients with cholangiocarcinoma admitted to the Hunan Provincial
People's Hospital between January 2016 and December 2016 were
recruited for the present study. The diagnosis for
cholangiocarcinoma was established by combining clinical symptoms
and examination with ultrasound, computed tomography, and magnetic
resonance imaging. Fifteen sex- and age-matched healthy individuals
were also recruited as controls. Venous blood was collected from
each participant (total 15 patients), and the serum concentration
of leptin (cat. no. RAB0333; Sigma-Aldrich; Merck KGaA) and plasma
concentration of PKM2 (cat. no. LS-F29732-1; LifeSpan BioSciences,
Inc.) were measured by ELISA kit, following the manufacturer's
protocols.
Statistical analysis
All data were analysed using SPSS 13.0 software
(SPSS, Inc.) and presented as the mean ± standard deviation. The
difference between two groups was analysed using an unpaired
two-tailed Student's t-test. One-way analysis of variance was used
for comparison among multiple groups and multiple comparisons were
further performed using the post hoc Tukey's test. P≤0.05 was
considered to indicate a statistically significant difference.
Results
Leptin induces EMT in cholangiocarcinoma
cells
Accumulating evidence suggests that leptin is a
pleiotropic cytokine involved in cancer development (23,24).
However, in cholangiocarcinoma, few studies have investigated the
functions or molecular mechanisms of leptin (19). To examine the biological
significance of leptin, its biosafety and effect on the
proliferation of the human TFK-1 cholangiocarcinoma cell line was
first assessed using an MTT assay. As shown in Fig. 1A, no significant decreases in cell
viability were observed when the cells were treated with 100 or 200
ng/ml leptin for 24 h, and leptin at 200 ng/ml promoted TFK-1 cell
proliferation when detected at 48 h, suggesting that leptin is not
toxic to TFK-1 cells. Next, the effect of leptin on the migratory
and invasive behaviours of TFK-1 cells was examined. In response to
increasing concentrations of leptin (0, 100 and 200 ng/ml), leptin
significantly stimulated the migration and the invasion of cells in
a dose-dependent manner, as indicated by Transwell assay (Fig. 1B and C). Monitoring the changes in
cell morphology revealed that leptin transformed the
cholangiocarcinoma cells from cuboidal shaped cells to elongated
spindle fibroblast-like cells (Fig.
1D), resembling the changes associated with EMT. To follow up,
the levels of several EMT promoters, including E-cadherin,
β-catenin, N-cadherin and vimentin were measured in cell lines
following leptin treatment. As shown in Fig. 1E and F, leptin treatment
significantly reduced the protein expression of epithelial markers
E-cadherin and β-catenin, while enhancing that of mesenchymal
markers N-cadherin and vimentin in a dose-dependent manner.
Collectively, these data support that leptin promotes EMT in
cholangiocarcinoma cells.
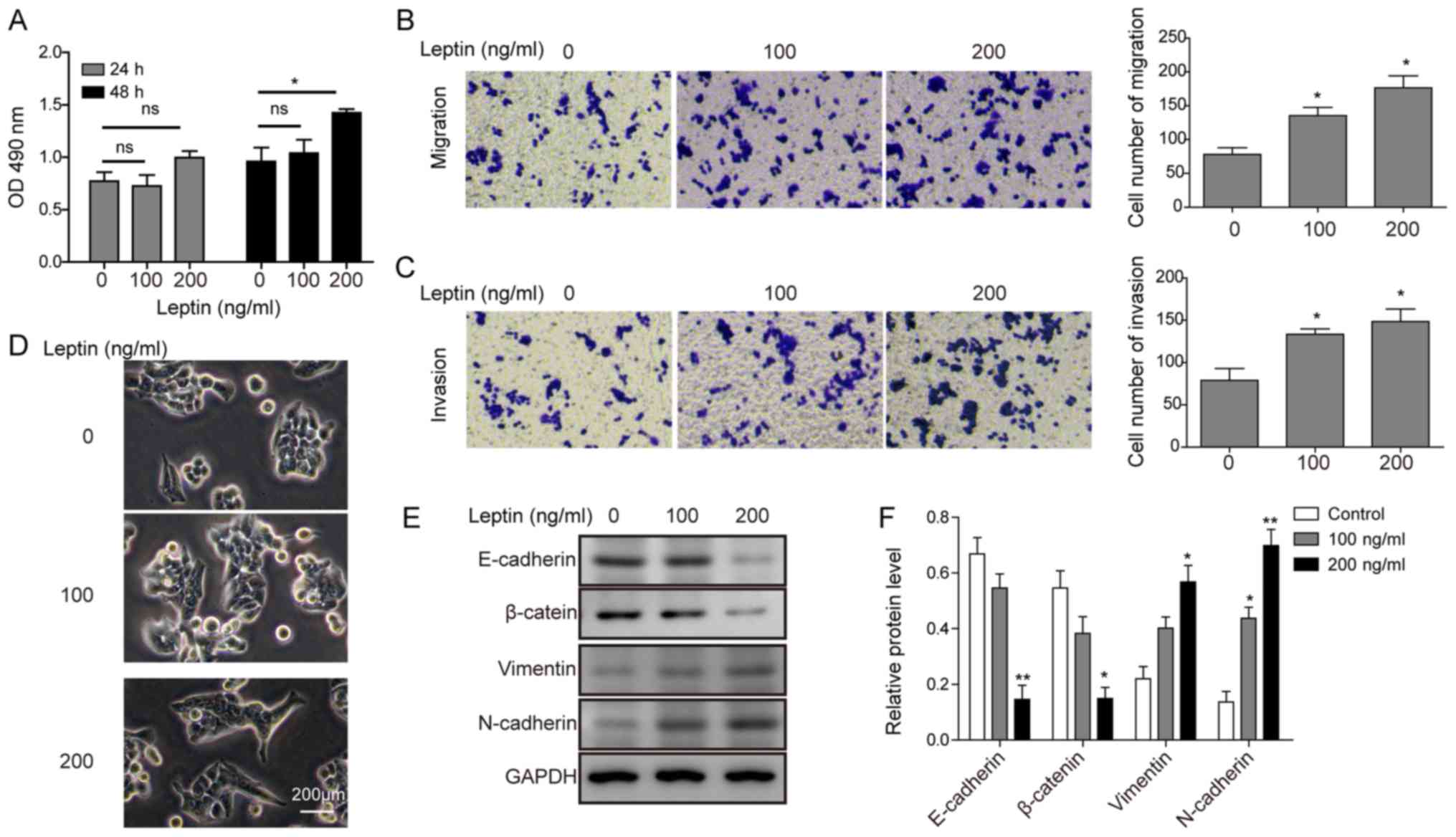 | Figure 1Leptin induces EMT in
cholangiocarcinoma cells. (A) TFK-1 cells were exposed to different
treatments for 24 or 48 h, and cell viability was detected by MTT
assay. (B) TFK-1 cell migration and (C) invasion were examined with
leptin treatment at the indicated concentrations. After 24 h, the
migrated or invaded cells were stained with crystal violet, imaged
(left panels), and counted under the microscope (right panel). Each
condition was tested in triplicate. (D) Morphological changes of
TFK-1 cells following treatment with 100 and 200 ng/ml leptin.
Scale bar, 200 μm. (E) TFK-1 cells were treated with leptin
at the indicated concentrations for 24 h. Protein expression levels
of E-cadherin, β-catenin, N-cadherin, and vimentin were examined by
western blot analysis. GAPDH was detected as the internal control.
(F) Quantification of protein levels (relative to GAPDH) from D.
Data are presented as the mean ± standard deviation from at least
three independent experiments. *P<0.05 and
**P<0.01. EMT, epithelial-mesenchymal transition; OD,
optical density; ns, not significant. |
Leptin stimulates the pro-angiogenic
capability of cholangiocarcinoma cells
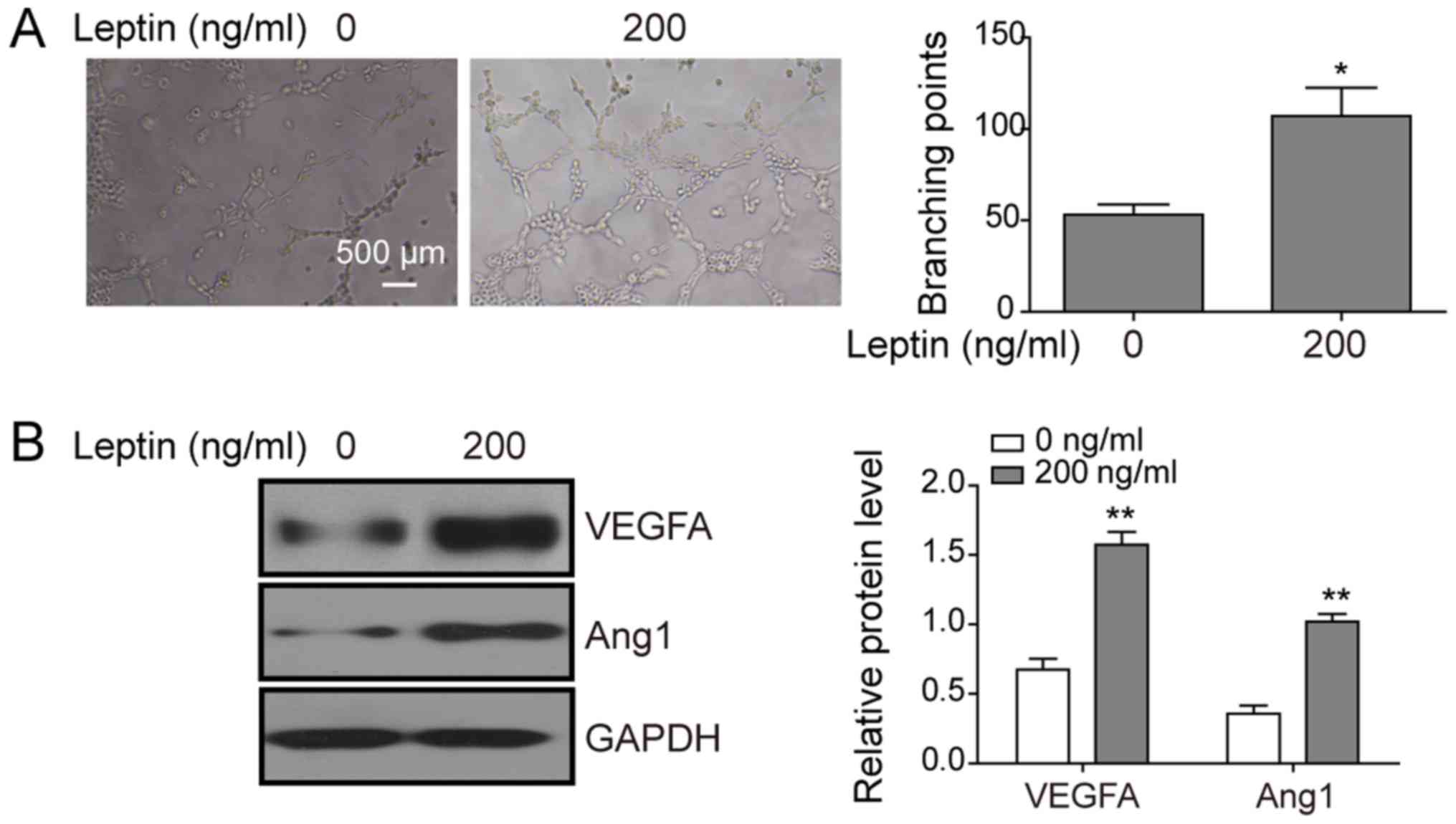
To examine the activity of leptin in angiogenesis,
TFK-1 cells were treated with 0 or 100 ng/ml leptin and the
conditioned medium was collected from these cells. By applying the
conditioned medium to HUVECs, monitoring their angiogenesis
(Fig. 2A, left), and measuring the
number of branching points (Fig.
2A, right), conditioned medium from leptin-treated
cholangiocarcinoma cells was demonstrated to induce tubular
formation of HUVECs. Concomitantly, leptin significantly increased
the protein expression of VEGFA and Ang1 (Fig. 2B), which are known pro-angiogenic
factors in cholangiocarcinoma cells, suggesting that leptin
stimulates the pro-angiogenic capability of cholangiocarcinoma
cells.
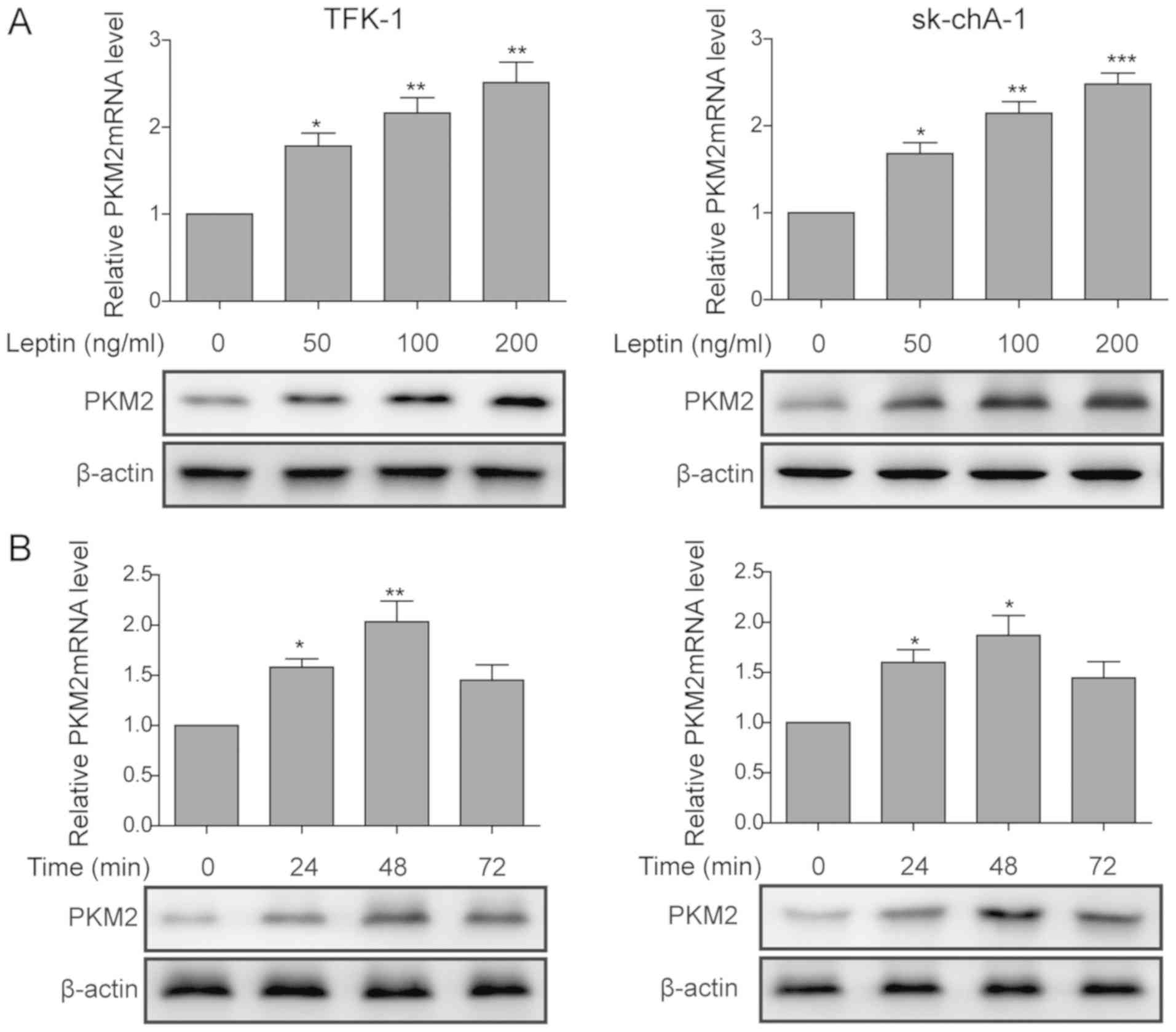
Leptin increases the expression of PKM2
in cholangiocarcinoma cells
To explore the molecular mechanisms underlying the
EMT- and angiogenesis-stimulating activities of leptin, PKM2, a
pyruvate kinase essential for leptin-induced EMT in breast cancer,
was examined (15). By measuring
PKM2 on the steady-state mRNA and the protein level in TFK-1 and
sk-chA-1 cells, leptin was revealed to significantly upregulate the
expression of PKM2 in a dose-dependent manner (Fig. 3A). The time-course study revealed
that PKM2 mRNA and protein (Fig.
3B) peaked at 24 h after leptin treatment, and gradually
reduced thereafter in TFK-1 and sk-chA-1 cells. Taken together,
these results suggest that leptin increased the expression of PKM2
in cholangiocarcinoma cells.
PKM2 is essential for leptin-induced EMT
and pro-angiogenesis
To assess the significance of upregulated PKM2 in
leptin-mediated EMT or pro-angiogenesis of cholangiocarcinoma
cells, the expression of endogenous PKM2 was targeted with siRNA
(si-PKM2). RT-qPCR and western blot analysis demonstrated that
si-PKM2 significantly and specifically knocked down the level of
PKM2 in TFK-1 cells (Fig. 4A).
When compared with vehicle control (PBS only) treatment, leptin
alone or leptin together with si-NC, si-PKM2 completely abolished
the effects of leptin on cell migration (Fig. 4B), invasion (Fig. 4C), and mesenchymal morphology
(Fig. 4D). Then, the expression of
EMT markers E-cadherin, β-catenin, N-cadherin, and vimentin were
detected by western blot analysis, which demonstrated that si-PKM2
abolished the effects of leptin (Fig.
4E). Furthermore, conditioned medium from si-PKM2-treated
cholangiocarcinoma cells abolished tubular formation of HUVECs
(Fig. 4F) and inhibited the
expression of VEGFA or Ang1 (Fig.
4G) in cholangiocarcinoma cells. Taken together, these data
suggest that PKM2 is an essential signalling molecule for
leptin-induced EMT and pro-angiogenesis of cholangiocarcinoma
cells.
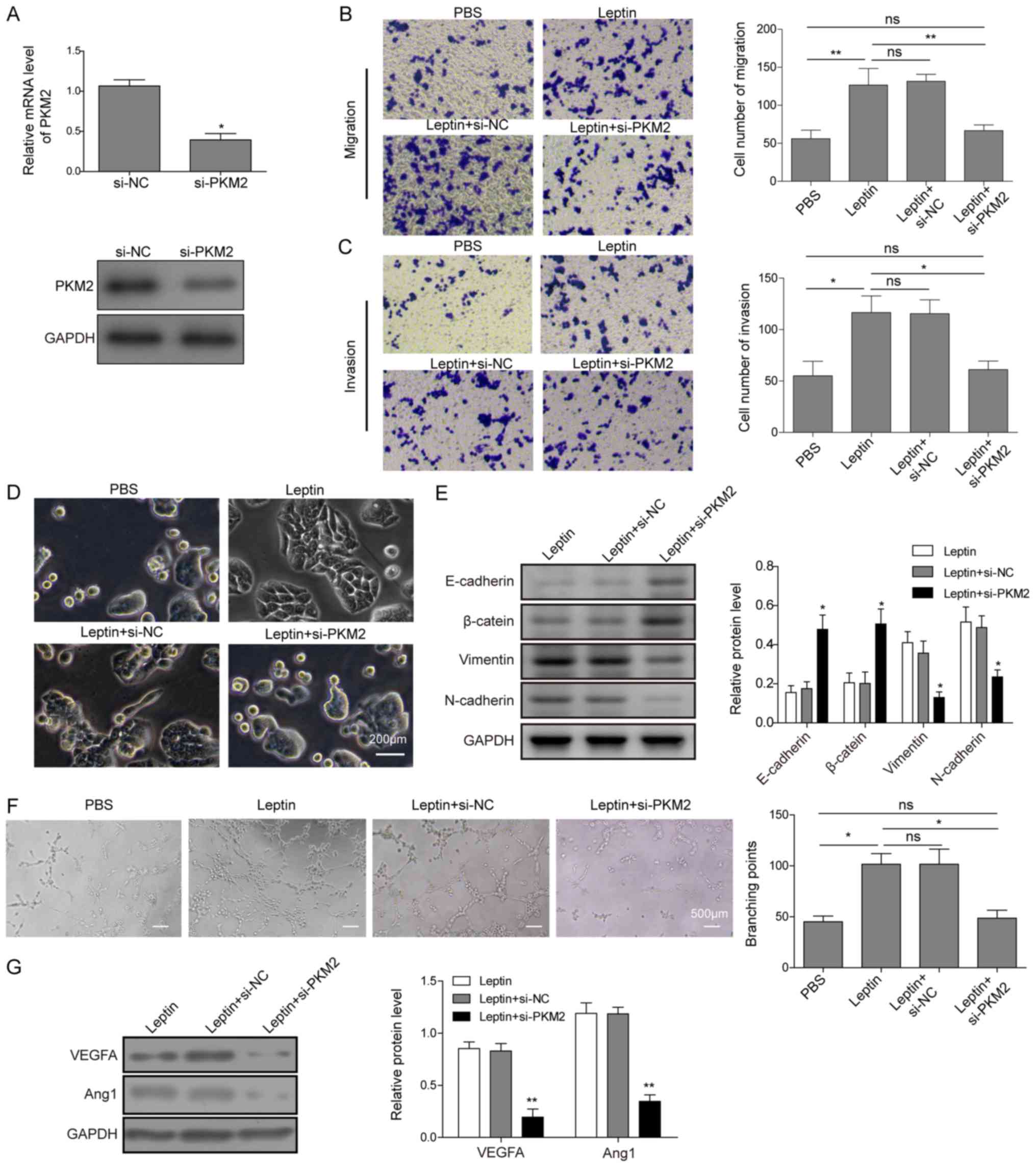 | Figure 4PKM2 is essential for leptin-induced
EMT and angiogenesis. (A) TFK-1 cells were transfected with either
negative control siRNA (si-NC) or si-PKM2, and the expression of
PKM2 was examined by reverse transcription-quantitative PCR and
western blot analysis. (B) TFK-1 cells not transfected, transfected
with negative control siRNA (si-NC), or si-PKM2 were treated with
vehicle control (PBS) or with leptin (100 ng/ml) for 24 h.
Migration and (C) invasion were examined by Transwell assays in
triplicate, with representative images of migrated or invaded cells
presented on the left and the quantification on the right. (D) Cell
morphology was imaged under a light microscope. Scale bar, 200
μm. (E) Expression of E-cadherin, β-catenin, N-cadherin, and
vimentin was examined by western blot analysis. GAPDH was detected
as the internal control (left panels). Quantification of protein
levels (relative to GAPDH) is presented (right panels). (F)
Cholangiocarcinoma cells not transfected, and transfected with
negative control siRNA (si-NC) or si-PKM2 were treated with leptin
(100 ng/ml), and the conditioned medium was collected. The
tubulogenesis assay was performed on HUVECs growing on Matrigel
upon treatment with the conditioned medium. Representative images
from each group are presented on the left and the quantification of
branching points on the right. Scale bar, 500 μm. (G)
Expression of VEGFA and Ang1 in indicated cholangiocarcinoma cells
was examined by western blot analysis, with representative images
presented on the left and the quantification of relative protein
levels on the right. GAPDH was detected as the internal control.
Data are presented as the mean ± standard deviation from at least
three independent experiments. *P<0.05 and
**P<0.01. PKM2, pyruvate kinase muscle isozyme M2;
EMT, epithelial-mesenchymal transition; si, small interfering;
HUVECs, human umbilical vein endothelial cells; VEGFA, vascular
endothelial growth factor A; Ang1, angiopoietin 1; ns, not
significant. |
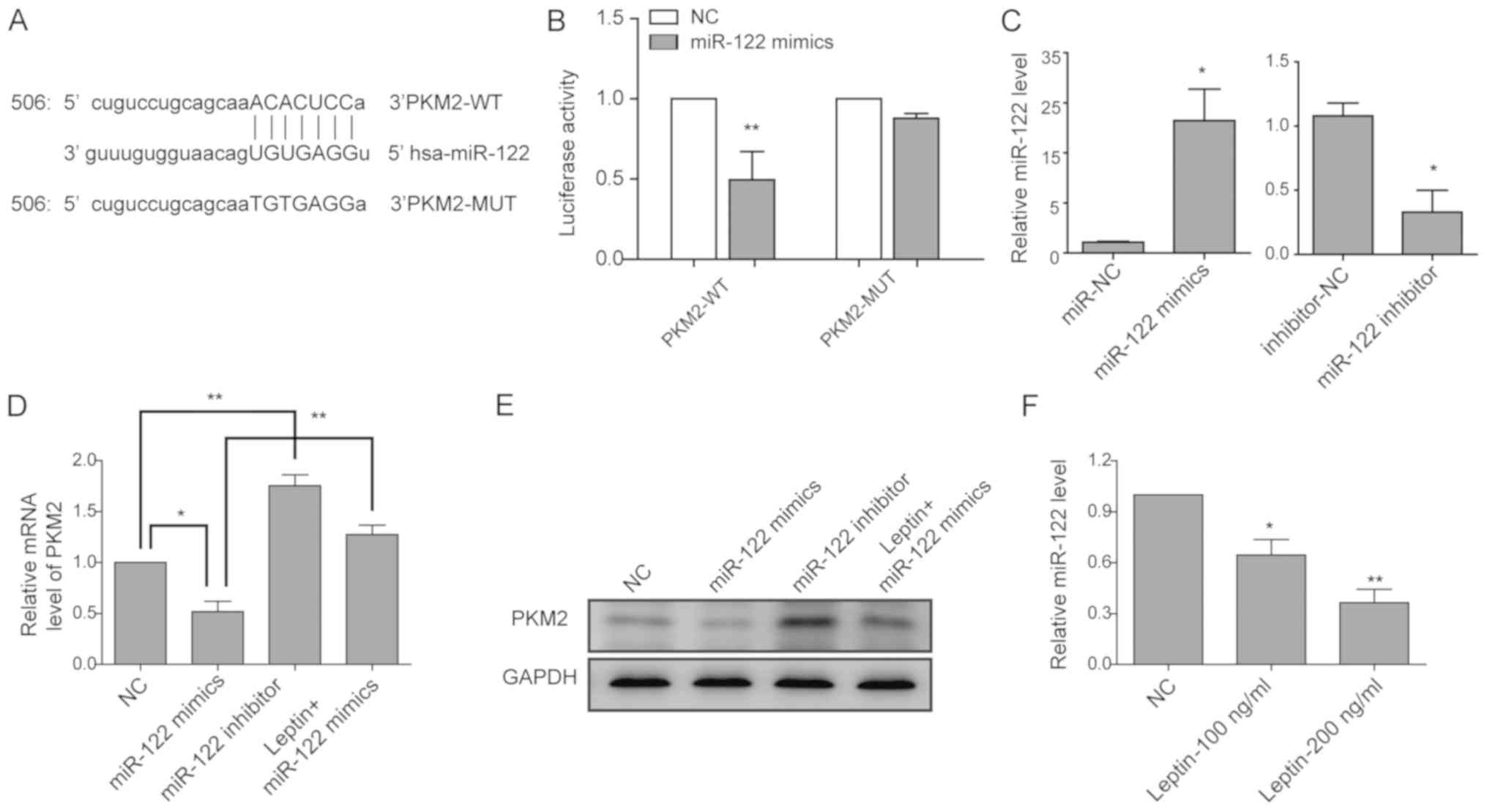
miR-122 mediates the regulation of leptin
on PKM2
To identify the molecules controlling
leptin-mediated regulation on PKM2, a bioinformatics search was
performed, and miR-122 was predicted as a potential miRNA that
interacts with the 3′-UTR of PKM2 (Fig. 5A). The luciferase reporter assay
demonstrated that miR-122 mimics significantly reduced the
luciferase activity of the reporter carrying the 3′-UTR of PKM2. In
contrast, a mutated luciferase reporter was developed by
site-directed mutagenesis, and the results suggested that mutations
on the binding sites successfully abolished the suppressive effect
of miR-122 (Fig. 5B). Next,
miR-122 mimics and miR-122 inhibitors were used for
gain-of-function and loss-of-function experiments, respectively.
RT-qPCR analysis revealed that miR-122 mimics elevated the miR-122
level to ~23-fold of that in cells transfected with miR-NC, while
miR-122 inhibitor significantly reduced the level of miR-122 when
compared with the expression in cells transfected with inhibitor NC
(Fig. 5C). Consistently, miR-122
mimics significantly reduced, while miR-122 inhibitors potently
increased the endogenous mRNA (Fig.
5D) as well as the protein level of PKM2 (Fig. 5E) in cholangiocarcinoma cells. The
addition of leptin reduced the inhibitory effect of miR-122 mimics
on PKM2 expression (Fig. 5D and
E). Furthermore, leptin significantly reduced the miR-122 level
in a dose-dependent manner (Fig.
5F). Collectively, these data suggest that leptin may target
miR-122, thus releasing miR-122-mediated inhibition on PKM2.
miR-122 mimics reverse leptin-induced EMT
and are pro-angiogenesis
To analyse the functional significance of miR-122 as
a negative regulator for leptin-mediated expression of PKM2,
cholangiocarcinoma cells were treated with leptin together with
miR-122 mimics. As shown in Fig. 6A
and B, miR-122 mimics were sufficient to reduce leptin-induced
migration and invasion of cholangiocarcinoma cells to the basal
level (when cells were treated with PBS) and a level significantly
lower compared with that in cells treated with leptin alone or
leptin with vehicle control (leptin+NC). In addition, miR-122
mimics significantly suppressed the changes in cell morphology
(Fig. 6C) and gene expression
(Fig. 6D) associated with
leptin-induced EMT. The conditioned medium collected from
cholangiocarcinoma cells treated with leptin+miR-122 mimics
presented similar activities as the medium from cells treated with
leptin+si-PKM2 (Fig. 5E),
disrupting tubular formation of HUVECs (Fig. 6E), which was associated with the
reduced expression of VEGFA and Ang1 (Fig. 6F). Taken together, these results
suggested that increasing miR-122 activity is sufficient to reverse
leptin-induced EMT as well as the pro-angiogenic capability of
cholangiocarcinoma cells.
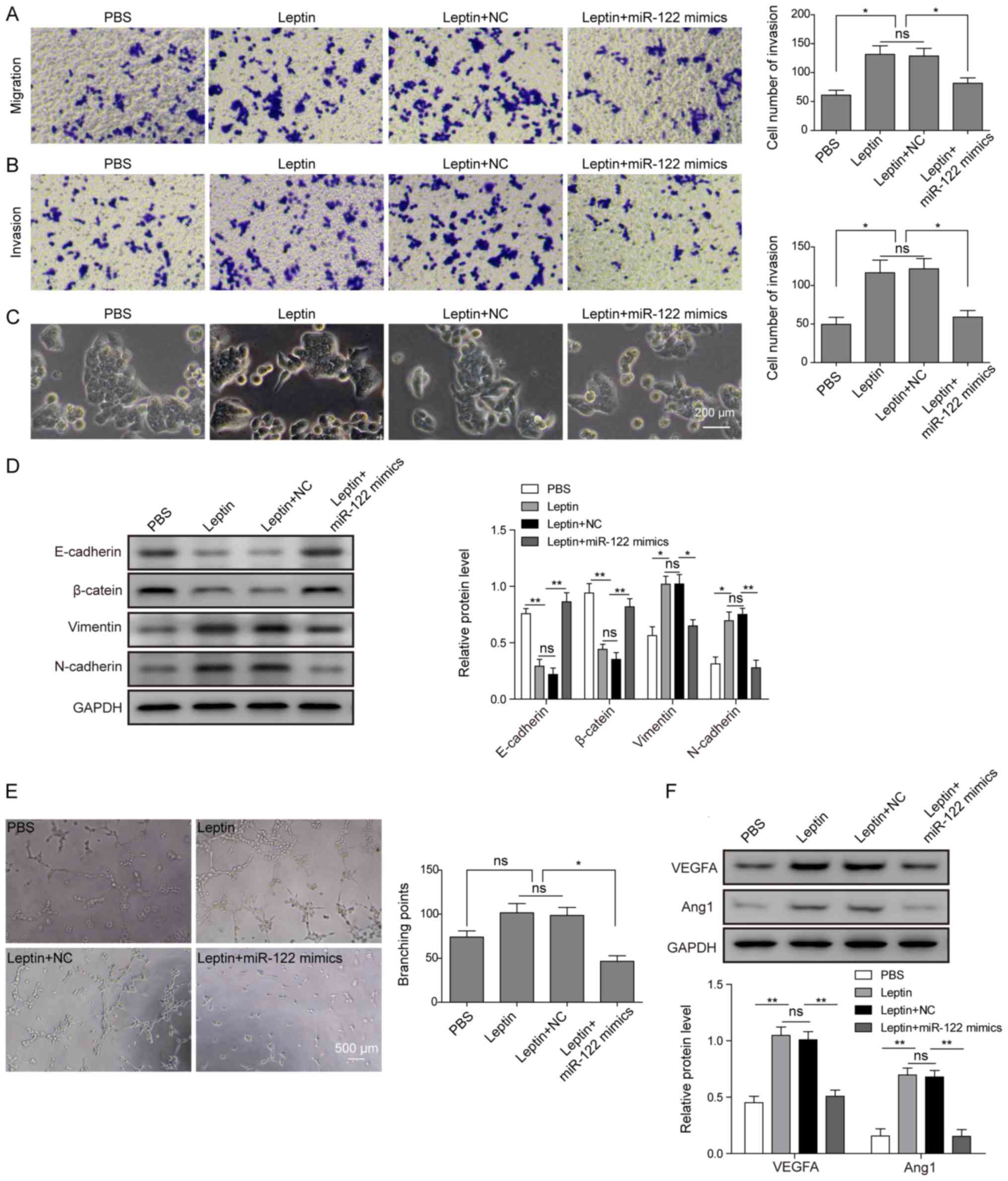 | Figure 6miR-122 mimics reverses
leptin-induced EMT and angiogenesis. (A) TFK-1 cells were treated
with PBS, leptin (100 ng/ml), leptin+NC, or leptin+miR-122 mimics.
Cell migration and (B) invasion were examined by Transwell assays
in triplicate, with representative images of migrated or invaded
cells presented on the left and the quantification on the right.
(C) Cell morphology was imaged under a light microscope. Scale bar,
200 μm. (D) Expression of E-cadherin, β-catenin, N-cadherin,
and vimentin was examined by western blot analysis. GAPDH was
detected as the internal control. (E) Cholangiocarcinoma cells were
treated with PBS, leptin (100 ng/ml), leptin+NC, or leptin+miR-122
mimics, and the conditioned medium was collected. The tube
formation assay was performed on HUVECs growing on Matrigel upon
treatment with the conditioned medium. Representative images from
each group are presented on the left and the quantification of
branching points on the right. Scale bar, 500 μm. (F)
Expression of VEGFA and Ang1 in cholangiocarcinoma cells was
examined by western blot analysis. Representative images and
quantification of relative protein levels are shown. GAPDH was
detected as the internal control. Data are presented as the mean ±
standard deviation from at least three independent experiments.
*P<0.05 and **P<0.01. EMT,
epithelial-mesenchymal transition; NC, negative control; HUVECs,
human umbilical vein endothelial cells; VEGFA, vascular endothelial
growth factor A; Ang1, angiopoietin 1. |
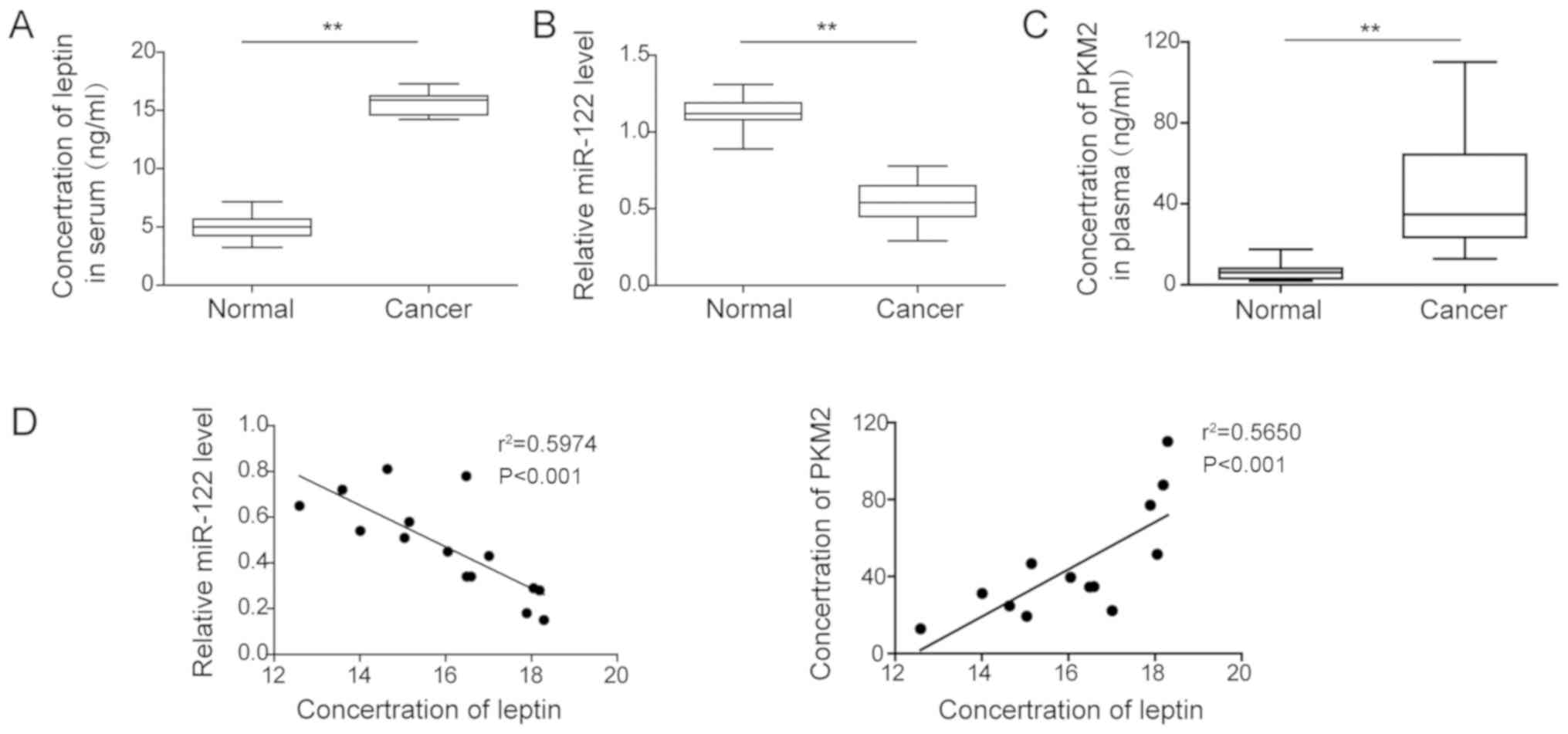
Serum level of leptin is positively
correlated with PKM2, but negatively correlated with miR-122 in
cholangiocarcinoma patients
The data presented above support the essential role
of the leptin/miR-122/PKM2 axis in regulating EMT and angiogenesis
of cholangiocarcinoma. To analyze the significance of this axis in
the clinical setting, the serum levels of leptin and miR-122, and
plasma level of PKM2 from patients with cholangiocarcinoma and age-
and sex-matched healthy individuals were measured. As shown in
Fig. 7A-C, serum leptin and PKM2
levels were significantly higher, while that of miR-122 was
significantly reduced in patients when compared with the
corresponding levels in healthy controls. Further correlation
analysis revealed that in the patients, the serum level of leptin
positively correlated with that of PKM2, but negatively with that
of miR-122 (Fig. 7D).
Discussion
In the present study, it was demonstrated that
leptin is a potent cytokine capable of inducing EMT and stimulating
the pro-angiogenic capability of cholangiocarcinoma cells.
Furthermore, the effects of leptin on cholangiocarcinoma cells were
indicated to be mediated through the leptin/miR-122/PKM2 axis as
the upregulation of miR-122 or downregulation of PKM2 was
sufficient to inhibit leptin-induced phenotypes. Consistently, the
serum level of leptin was significantly higher in patients with
cholangiocarcinoma compared with healthy controls, and positively
correlated with serum PKM2 levels, but negatively correlated with
serum miR-122 levels. The results of the present study support the
notion that leptin is an ideal therapeutic target for
cholangiocarcinoma since antagonizing leptin was able to
simultaneously target multiple phenotypes responsible for the
malignant progression of the disease.
An increasing body of evidence supports the
association between obesity and a higher risk of developing
different types of cancer (25,26).
In a study on 30 patients with colorectal, breast, lung, testicular
or gastric cancer, no significant differences were observed in the
serum leptin level at baseline or in response to chemotherapy
between patients and matched healthy controls, although leptin was
identified to be positively associated with the body mass index of
patients (27). By contrast, when
focusing on a specific type of cancer, several studies suggest a
positive association between higher serum leptin level and cancers
(28,29), while some suggest a negative
correlation (11,30). Meta-analysis revealed that obesity
is a risk factor for cholangiocarcinoma (31). Consistently, the results of the
present study demonstrated that the baseline level of serum leptin
was significantly higher in cholangiocarcinoma patients compared
with that in healthy individuals, indicating that leptin is
functionally important for the development of
cholangiocarcinoma.
Aggressive development and early metastatic spread
constitute the major obstacles for effective treatment of
cholangiocarcinoma. Therefore, understanding and targeting
mechanisms responsible for the malignancy of the disease may aid in
developing novel therapies for cholangiocarcinoma. Previous studies
have presented convincing evidence that EMT is present in
cholangiocarcinoma development and progression, but also associated
with poor prognosis of patients (32,33).
In the present study, leptin was revealed to be functionally
capable of inducing EMT phenotypes in cholangiocarcinoma cells:
Stimulating their migration and invasion; changing tumour cells
from the cuboidal epithelial morphology to a spindle mesenchymal
shape; downregulating junction molecules, E-cadherin and β-catenin;
and upregulating mesenchymal markers, N-catenin and vimentin.
Although, to the best of our knowledge, this is the first study to
demonstrate that leptin is capable of inducing EMT in
cholangiocarcinoma, EMT is not a novel functional phenotype for
leptin, as has been well documented in multiple cancer types,
including breast (34), lung
(35), and ovarian (36) cancer. Several signalling pathways
have been identified to mediate leptin-induced EMT, including
PI3K/AKT/PKM2 (15), TGF-β
(35), AKT/GSK3β and MTA1/Wnt1
axes (34). In the present study,
leptin increased the expression of PKM2 in cholangiocarcinoma
cells, and siRNA-mediated downregulation of PKM2 was sufficient to
abolish multiple leptin-induced EMT phenotypes.
Notably, through bioinformatic miRNA screening,
miR-122 was identified as a negative regulator between leptin and
PKM2, whereby leptin, through the suppression of miR-122, reversed
the inhibitory effects of miR-122 on PKM2 in cholangiocarcinoma
cells. The essential role of miR-122 in leptin-PKM2 regulation was
further corroborated by the findings that miR-122 mimics were
sufficient to downregulate endogenous PKM2, while miR-122
inhibitors significantly increased the expression of PKM2.
Karakatsanis et al (37)
reported the downregulation of miR-122 in intrahepatic
cholangiocarcinoma. Wu et al (38) demonstrated that, by inhibiting p53
expression, miR-122 was necessary and sufficient to reduce the
proliferation, and suppress the migration and invasion of
cholangiocarcinoma cells. Although these studies support the idea
that miR-122 functions as a tumour suppressor, they did not explore
the mechanism by which miR-122 inhibited the aggressive and
metastatic behaviours, specifically EMT of cholangiocarcinoma.
In addition to tumour cells, other mesenchymal
components within the tumour microenvironment, including
endothelial cells, fibroblasts, and macrophages are also essential
for the malignant progression of cholangiocarcinoma (39). Studies have established leptin as a
potent angiogenic factor and revealed multiple underlying
mechanisms: Activating P38 MAPK/AKT/COX-2 signalling or JAK/STAT
and AKT signalling pathways; and increasing the production of VEGF,
fibroblast growth factor-2 (FGF-2) and matrix metalloproteinases
from endothelial cells (40). In
the present study, the leptin/miR-122/PKM2 axis was demonstrated to
regulate the pro-angiogenic capability of cholangiocarcinoma cells
in vitro. Conditioned medium from tumour cells treated with
miR-122 mimics and si-PKM2 presented comparable effects in
disrupting tubular formation associated with the downregulation of
VEGF and Ang1 expression in cholangiocarcinoma cells. Although Wang
et al (41) suggested that
miR-122 directly targeted VEGF through post-transcriptional
regulation, the findings of the present study indicate that it may
indirectly target VEGF expression via PKM2. Consistently, Azoitei
et al (42) demonstrated
that PKM2 promoted HIF-1α accumulation and stimulated the
subsequent secretion of VEGF from pancreatic cancer cells by
activating NF-κB. Li et al (43) reported that PKM2 increased
endothelial cell proliferation, migration and adhesion to the
extracellular matrix. As the angiogenesis assays were performed on
endothelial cells using the conditioned medium from
cholangiocarcinoma cells in the present study, the possibility that
the leptin/miR-122/PKM2 axis may directly act on endothelial cells
to stimulate angiogenesis in an autocrine manner cannot be
excluded.
In conclusion, the results of the present study
provided clinical relevance and demonstrated the pleiotropic
activities of leptin in promoting the malignant transformation of
cholangiocarcinoma. Leptin not only stimulates multiple EMT
phenotypes, but also enhances the pro-angiogenic activity of cancer
cells. Therefore, targeting leptin may aid in supressing a
multitude of malignant behaviours of cholangiocarcinoma and achieve
superior anticancer effects. Molecularly, the actions of leptin in
cholangiocarcinoma cells appear to be mediated through the
miR-122/PKM2 axis, allowing for the development of novel anticancer
therapies that enable the fine-tuning of leptin activities. As this
study was primarily performed using cells cultured in vitro,
it is essential to follow up this study with properly established
in vivo cholangiocarcinoma models.
Funding
The present study was supported by the Scientific
Research Project of Hunan Provincial Education Department (grant
no. 17C0963), the Research Foundation of Hunan Provincial Health
Bureau (grant no. B2007136) and the Science and Technology Program
of Hunan Provincial Science and Technology Department (grant no.
2009SK3079).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
BJ substantially contributed to the conception of
this study, as well as edited the manuscript. CP was involved in
the study conception and design. ZS performed the literature
research and experimental studies. WY performed the clinical
studies. OL performed data acquisition and edited the manuscript.
ZT and BJ performed data and statistical analysis. CG was involved
in drafting the manuscript and revising it critically for important
intellectual content. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Board of Hunan Provincial People's Hospital, and written
informed consent was obtained from all participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Patel T: Cholangiocarcinoma -
controversies and challenges. Nat Rev Gastroenterol Hepatol.
8:189–200. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Anderson CD, Pinson CW, Berlin J and Chari
RS: Diagnosis and treatment of cholangiocarcinoma. Oncologist.
9:43–57. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gupta GP and Massague J: Cancer
metastasis: Building a framework. Cell. 127:679–695. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guarino M, Rubino B and Ballabio G: The
role of epithelial-mesenchymal transition in cancer pathology.
Pathology. 39:305–318. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vaquero J, Guedj N, Clapéron A, Nguyen
Ho-Bouldoires TH, Paradis V and Fouassier L: Epithelial-mesenchymal
transition in cholangiocarcinoma: From clinical evidence to
regulatory networks. J Hepatol. 66:424–441. 2017. View Article : Google Scholar
|
|
7
|
Heerboth S, Housman G, Leary M, Longacre
M, Byler S, Lapinska K, Willbanks A and Sarkar S: EMT and tumor
metastasis. Clin Transl Med. 4:62015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kelesidis T, Kelesidis I, Chou S and
Mantzoros CS: Narrative review: the role of leptin in human
physiology: emerging clinical applications. Ann Intern Med.
152:93–100. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mohammadzadeh G, Ghaffari MA, Bafandeh A
and Hosseini SM: Association of serum soluble leptin receptor and
leptin levels with breast cancer. J Res Med Sci. 19:433–438.
2014.PubMed/NCBI
|
|
10
|
Karapanagiotou EM, Tsochatzis EA, Dilana
KD, Tourkantonis I, Gratsias I and Syrigos KN: The significance of
leptin, adiponectin, and resistin serum levels in non-small cell
lung cancer (NSCLC). Lung Cancer. 61:391–397. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brown DR, Berkowitz DE and Breslow MJ:
Weight loss is not associated with hyperleptinemia in humans with
pancreatic cancer. J Clin Endocrinol Metab. 86:162–166. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wallace AM, Sattar N and McMillan DC:
Effect of weight loss and the inflammatory response on leptin
concentrations in gastrointestinal cancer patients. Clin Cancer
Res. 4:2977–2979. 1998.PubMed/NCBI
|
|
13
|
Dutta D, Ghosh S, Pandit K, Mukhopadhyay P
and Chowdhury S: Leptin and cancer: Pathogenesis and modulation.
Indian J Endocrinol Metab. 16(Suppl 3): S596–S600. 2012. View Article : Google Scholar
|
|
14
|
Morris DL and Rui L: Recent advances in
understanding leptin signaling and leptin resistance. Am J Physiol
Endocrinol Metab. 297:E1247–E1259. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wei L, Li K, Pang X, Guo B, Su M, Huang Y,
Wang N, Ji F, Zhong C, Yang J, et al: Leptin promotes
epithelial-mesenchymal transition of breast cancer via the
upregulation of pyruvate kinase M2. J Exp Clin Cancer Res.
35:1662016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Christofk HR, Vander Heiden MG, Harris MH,
Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL and
Cantley LC: The M2 splice isoform of pyruvate kinase is important
for cancer metabolism and tumour growth. Nature. 452:230–233. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hamabe A, Konno M, Tanuma N, Shima H,
Tsunekuni K, Kawamoto K, Nishida N, Koseki J, Mimori K, Gotoh N, et
al: Role of pyruvate kinase M2 in transcriptional regulation
leading to epithelial-mesenchymal transition. Proc Natl Acad Sci
USA. 111:15526–15531. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang WH, Chen JC, Hsu KH, Lin CY, Wang SW,
Wang SJ, Chang YS and Tang CH: Leptin increases VEGF expression and
enhances angiogenesis in human chondrosarcoma cells. Biochim
Biophys Acta. 1840:3483–3493. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fava G, Alpini G, Rychlicki C, Saccomanno
S, DeMorrow S, Trozzi L, Candelaresi C, Venter J, Di Sario A,
Marzioni M, et al: Leptin enhances cholangiocarcinoma cell growth.
Cancer Res. 68:6752–6761. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Wong N and Wang X: miRDB: An online
resource for microRNA target prediction and functional annotations.
Nucleic Acids Res. 43(D1): D146–D152. 2015. View Article : Google Scholar :
|
|
22
|
Li JH, Liu S, Zhou H, Qu LH and Yang JH:
starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA
interaction networks from large-scale CLIP-Seq data. Nucleic Acids
Res. 42(D1): D92–D97. 2014. View Article : Google Scholar
|
|
23
|
Candela r ia PV, Rampoldi A, Ha rbuza r iu
A and Gonzalez-Perez RR: Leptin signaling and cancer
chemoresistance: Perspectives. World J Clin Oncol. 8:106–119. 2017.
View Article : Google Scholar
|
|
24
|
Park J and Scherer PE: Leptin and cancer:
From cancer stem cells to metastasis. Endocr Relat Cancer.
18:C25–C29. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gao C, Patel CJ, Michailidou K, Peters U,
Gong J, Schildkraut J, Schumacher FR, Zheng W, Boffetta P, Stucker
I, et al the Colorectal Transdisciplinary Study (CORECT);
Discovery, Biology and Risk of Inherited Variants in Breast Cancer
(DRIVE); Elucidating Loci Involved in Prostate Cancer
Susceptibility (ELLIPSE); Follow-up of Ovarian Cancer Genetic
Association and Interaction Studies (FOCI); and Transdisciplinary
Research in Cancer of the Lung (TRICL): Mendelian randomization
study of adiposity-related traits and risk of breast, ovarian,
prostate, lung and colorectal cancer. Int J Epidemiol. 45:896–908.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kyrgiou M, Kalliala I, Markozannes, Gunter
MJ, Paraskevaidis E, Gabra H, Martin-Hirsch P and Tsilidis KK:
Adiposity and cancer at major anatomical sites: Umbrella review of
the literature. BMJ. 356:j4772017. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ayoub N, Alkhatatbeh M, Jibreel M and
Ababneh M: Analysis of circulating adipokines in patients newly
diagnosed with solid cancer: Associations with measures of
adiposity and tumor characteristics. Oncol Lett. 13:1974–1982.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Stattin P, Lukanova A, Biessy C, Söderberg
S, Palmqvist R, Kaaks R, Olsson T and Jellum E: Obesity and colon
cancer: Does leptin provide a link? Int J Cancer. 109:149–152.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang D, Gao L, Gong K, Chai Q and Wang G:
Increased serum leptin level in overweight patients with colon
carcinoma: A cross-sectional and prospective study. Mol Clin Oncol.
6:75–78. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bolukbas FF, Kilic H, Bolukbas C, Gumus M,
Horoz M, Turhal NS and Kavakli B: Serum leptin concentration and
advanced gastrointestinal cancers: A case controlled study. BMC
Cancer. 4:292004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li JS, Han TJ, Jing N, Li L, Zhang XH, Ma
FZ and Liu JY: Obesity and the risk of cholangiocarcinoma: A
meta-analysis. Tumour Biol. 35:6831–6838. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nitta T, Mitsuhashi T, Hatanaka Y,
Miyamoto M, Oba K, Tsuchikawa T, Suzuki Y, Hatanaka KC, Hirano S
and Matsuno Y: Prognostic significance of epithelial-mesenchymal
transition-related markers in extrahepatic cholangiocarcinoma:
Comprehensive immunohistochemical study using a tissue microarray.
Br J Cancer. 111:1363–1372. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Clapéron A, Mergey M, Nguyen Ho-Bouldoires
TH, Vignjevic D, Wendum D, Chrétien Y, Merabtene F, Frazao A,
Paradis V, Housset C, et al: EGF/EGFR axis contributes to the
progression of cholangiocarcinoma through the induction of an
epithelial-mesenchymal transition. J Hepatol. 61:325–332. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yan D, Avtanski D, Saxena NK and Sharma D:
Leptin-induced epithelial-mesenchymal transition in breast cancer
cells requires β-catenin activation via Akt/GSK3- and MTA1/Wnt1
protein-dependent pathways. J Biol Chem. 287:8598–8612. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Feng H, Liu Q, Zhang N, Zheng L, Sang M,
Feng J, Zhang J, Wu X and Shan B: Leptin promotes metastasis by
inducing an epithelial-mesenchymal transition in A549 lung cancer
cells. Oncol Res. 21:165–171. 2013. View Article : Google Scholar
|
|
36
|
Wei X, Liu Y, Gong C, Ji T, Zhou X, Zhang
T, Wan D, Xu S, Jin P, Yang X, et al: Targeting leptin as a
therapeutic strategy against ovarian cancer peritoneal metastasis.
Anticancer Agents Med Chem. 17:1093–1101. 2017. View Article : Google Scholar
|
|
37
|
Karakatsanis A, Papaconstantinou I,
Gazouli M, Lyberopoulou A, Polymeneas G and Voros D: Expression of
microRNAs, miR-21, miR-31, miR-122, miR-145, miR-146a, miR-200c,
miR-221, miR-222, and miR-223 in patients with hepatocellular
carcinoma or intrahepatic cholangiocarcinoma and its prognostic
significance. Mol Carcinog. 52:297–303. 2013. View Article : Google Scholar
|
|
38
|
Wu C, Zhang J, Cao X, Yang Q and Xia D:
Effect of mir-122 on human cholangiocarcinoma proliferation,
invasion, and apoptosis through P53 expression. Med Sci Monit.
22:2685–2690. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Leyva-Illades D, McMillin M, Quinn M and
Demorrow S: Cholangiocarcinoma pathogenesis: Role of the tumor
microenvironment. Transl Gastrointest Cancer. 1:71–80.
2012.PubMed/NCBI
|
|
40
|
Tahergorabi Z and Khazaei M: Leptin and
its cardiovascular effects: Focus on angiogenesis. Adv Biomed Res.
4:792015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang Y, Xing QF, Liu XQ, Guo ZJ, Li CY and
Sun G: miR-122 targets VEGFC in bladder cancer to inhibit tumor
growth and angiogenesis. Am J Transl Res. 8:3056–3066.
2016.PubMed/NCBI
|
|
42
|
Azoitei N, Becher A, Steinestel K, Rouhi
A, Diepold K, Genze F, Simmet T and Seufferlein T: PKM2 promotes
tumor angiogenesis by regulating HIF-1α through NF-κB activation.
Mol Cancer. 15:32016. View Article : Google Scholar
|
|
43
|
Li L, Zhang Y, Qiao J, Yang JJ and Liu ZR:
Pyruvate kinase M2 in blood circulation facilitates tumor growth by
promoting angiogenesis. J Biol Chem. 289:25812–25821. 2014.
View Article : Google Scholar : PubMed/NCBI
|