Introduction
Lung cancer is the leading cause of cancer-related
mortality worldwide. Over 85% of lung cancers are non-small-cell
lung cancers (NSCLCs), with adenocarcinoma being the most common
histological subtype of NSCLC. Despite continuous advances in the
diagnosis and treatment of NSCLC, ~90% of newly diagnosed patients
with advanced NSCLC succumb to the disease within 2 years. As the
rate of disseminated disease in such patients is high, numerous
attempts have been made to improve systemic treatment for over 2
decades (1).
Antrodia camphorata, a camphor tree mushroom,
is a valuable traditional Chinese herbal medicine that exhibits
pharmacological properties against several diseases. Antrodia
camphorata is rich in flavonoids, terpenoids, polyphenolics and
polysaccharides, and has been produced on an agricultural
manufacturing scale in Taiwan. The alcohol extract of Antrodia
camphorata had been found to exert anticancer effects through
inhibiting tumor cell DNA synthesis, promoting apoptosis and
exerting an antimigration effect (2).
Antroquinonol is a ubiquinone derivative isolated
from Antrodia camphorata. A previous study demonstrated that
antroquinonol displayed anticancer activity against hepatocellular
carcinoma cell lines through activation of 5′
adenosine-monophosphate-activated protein kinase and inhibition of
the mammalian target of rapamycin (mTOR) pathway (3). Further study demonstrated that
antroquinonol exhibits anticancer activity in human pancreatic
cancers through inhibition of the phosphoinositide-3 kinase
(PI3K)/Akt/mTOR pathway, which in turn downregulates the expression
of cell cycle regulators (4). The
translational inhibition causes a G1 arrest of the cell cycle and
ultimately mitochondria-dependent apoptosis. Moreover, autophagic
cell death and accelerated senescence may also explain the
antroquinonol-mediated anticancer effect. A study on the A549
pulmonary adenocarcinoma cell line demonstrated that
antroquinonol-induced apoptosis was associated with disrupted
mitochondrial membrane potential and activation of caspase-3 and
poly ADP ribose polymerase cleavage (5). Moreover, antroquinonol treatment
downregulated the expression of B-cell lymphoma 2 proteins, which
was correlated with decreased PI3K and mTOR protein levels, without
altering the levels of pro- or antiapoptotic proteins. The results
of the microarray analysis demonstrated that antroquinonol altered
the expression level of miRNAs in A549 cells compared with the
untreated control. These data collectively suggested that
antroquinonol exerted an antiproliferative effect on A549 cells
(5). In addition, antroquinonol
exhibited no observed genotoxicity or reproductive toxicity at
doses up to 80 mg/kg/day in the Ames test, mammalian cell gene
mutation test, micronucleus test, erythrocyte micronucleus test, or
in vivo study of Sprague Dawley rats (unpublished data).
Thus, this multicenter phase I trial was conducted
to determine the maximum tolerable dose (MTD) and evaluate the
pharmacokinetics (PKs), safety, tolerability and efficacy profiles
of antroquinonol use in metastatic NSCLC patients who had received
at least two prior systemic treatment regimens, including one
platinum-based chemotherapy regimen.
Patients and methods
Study design and treatment
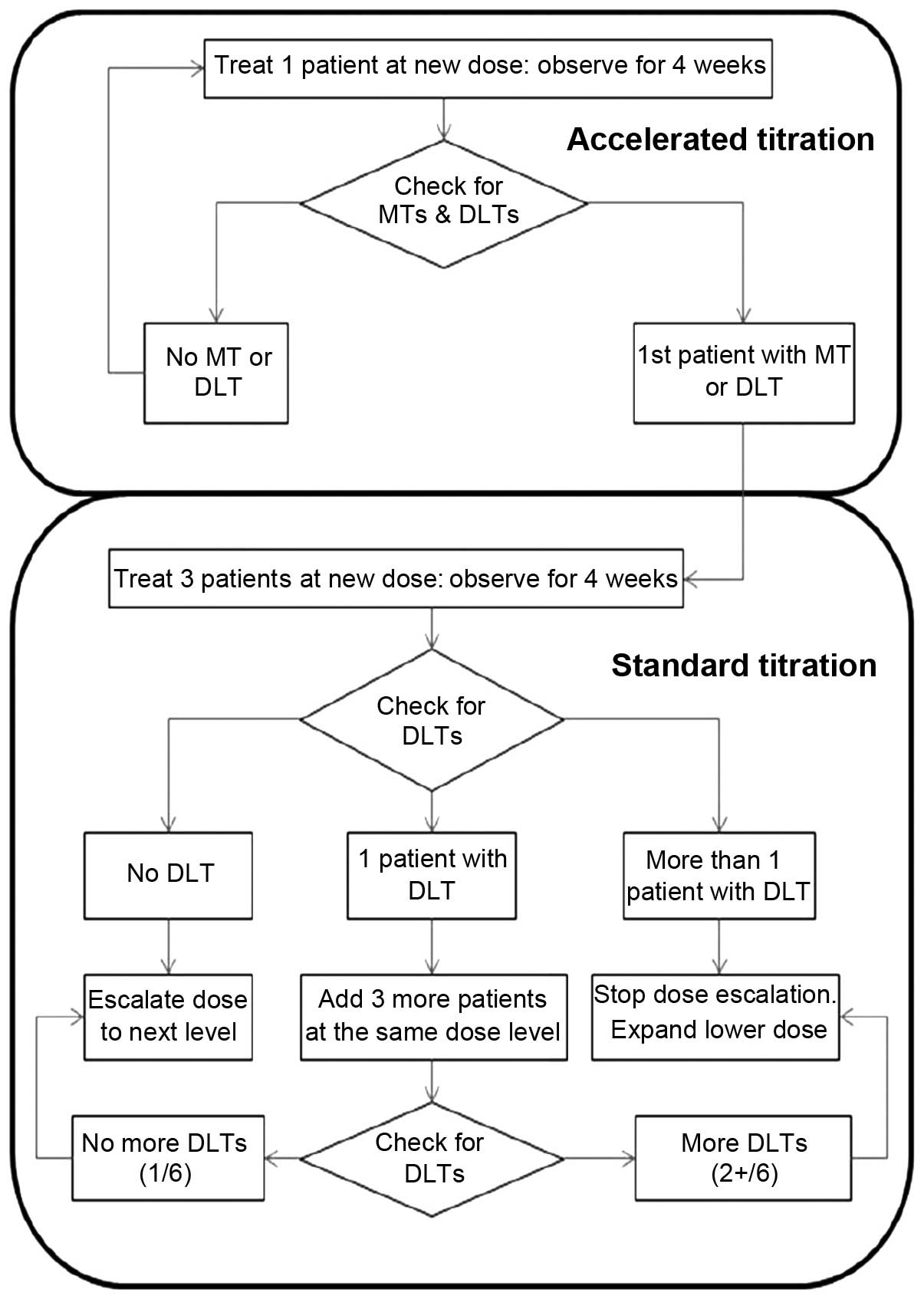
This was an open-label, non-randomized, dose
escalation PKs study conducted to determine the MTD and
dose-limiting toxicities (DLTs) and investigate the PKs, safety,
tolerability and preliminary efficacy profiles of antroquinonol. An
accelerated titration design was used for this study. For the 6
dose levels (daily dose of 50, 100, 200, 300, 450 and 600 mg), a
maximum of 36 patients were scheduled based on the criteria of a
maximum of 6 patients per cohort: 1 (lowest level) to 6 (highest
level) patients were scheduled for each dose group in the
accelerated titration phase, and 3–6 patients for each dose group
in the standard titration phase (6 dose cohorts) (Fig. 1). Antroquinonol was administered
per os daily, within 15 min after breakfast at the assigned
dose levels for 4 weeks. The study protocol was approved by the
Institutional Review Boards of Taipei Veterans General Hospital and
Tri-Service General Hospital (VGHIRB 201010007MB, TSGHIRB
099-01-007, clinicaltrials.gov protocol
registration system NCT011–340-16).
The primary endpoint was MTD. DLT was defined as any
≥grade 3 toxicity according to the National Cancer Institute Common
Terminology Criteria for Adverse Events (NCI CTCAE), version 4.03
(http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf),
as determined by the investigator, possibly related in terms of
causality to the administration of the study drug, and observed
during the first 4 weeks of treatment for patients in the
accelerated or standard titration phase. Grade ≥3 nausea, vomiting
and diarrhea were considered as DLTs only if they remained at grade
3 for >3 days despite adequate treatment. The secondary
endpoints included three parameters: PKs, preliminary efficacy
evaluation of changes in measurable tumor size, and safety
profile.
No other cytotoxic agents, targeted therapy,
investigational drugs, immunotherapy, or radiotherapy was permitted
during the study period. The following medications were also
prohibited: Levonorgestrel (internal standard in bioanalysis), and
drugs known to inhibit or induce cytochrome P (CYP)2C19, CYP3A4,
CYP2C8 and CYP2E1.
Patients
Patients aged ≥20 years with metastatic NSCLC of the
lung who had developed progressive disease after two lines of
chemotherapy (including one platinum-based) and one line of
epidermal growth factor receptor (EGFR)-targeted therapy (if the
patient harboured an EGFR mutation or if the EGFR mutation status
was unknown) were enrolled in this study, after providing written
informed consent. Other eligibility criteria included: A
histological or cytological diagnosis of adenocarcinoma of the lung
(or mixed-cell type tumor with an adenocarcinomatous component);
life expectancy of ≥3 months; Eastern Cooperative Oncology Group
performance status score of ≤2; recovery from toxicities of
previous anticancer treatments to ≤grade 1 NCI CTCAE, except for
alopecia; clinically measurable disease; no previous radiotherapy
directed at the measurable lesion(s); adequate bone marrow reserve,
with a white blood cell count of ≥3,500/mm3, hemoglobin
concentration of ≥9.0 g/dl, and platelet count of ≥100,000
cells/mm3; adequate liver function; and adequate renal
function.
The patients were categorized into an
intent-to-treat (ITT) population (patients who received at least
one dose of antroquinonol), a per-protocol (PP) population
[patients who completed at least 3 cycles of treatment with proper
imaging assessment according to Response Evaluation Criteria in
Solid Tumors (RECIST) (6)] and a PK
population (patients who received at least one dose of
antroquinonol with sufficient post-dose bio-samples collected for
PKs profile characterization). Baseline and demographic
characteristics and safety analyses were performed in the ITT
population. The PK population was used for PKs analyses.
PKs evaluation
For each dose cohort, samples were obtained from all
patients treated in the accelerated and standard titration phases
of the study. Blood sampling was performed on days 1, 14, 27 and 28
of the first treatment cycle only. Serial blood samples (5 ml per
sample) were collected for 24 h at the following time points on
study days 1 and 28: Day 1, within 30 min prior to and at 0.25,
0.5, 1, 2, 3, 4, 6, 8, 10, 14 and 24 h after dose administration;
and day 28, immediately prior to and at 0.25, 0.5, 1, 2, 3, 4, 6,
8, 10, 14 and 24 h after dose administration. A blood sample for
trough plasma concentration was collected immediately prior to dose
administration on days 14 and 27.
Determining the plasma PKs of antroquinonol involved
investigating the following parameters during a maximum of 4 weeks
of dose administration in the accelerated and standard titration
phases: Cmax day1, observed maximum plasma concentration
after dosing on day 1; Tmax day1, time to reach
Cmax day1 on day 1; AUC0-t day1, truncated
area under the plasma concentration-time curve from the beginning
of dosing to the last measurable concentration on day 1; T1/2
day1, half-life on day 1; Cmax day28, observed
maximum plasma concentration after dosing on day 28; Tmax
day28, time to reach Cmax day28 on day 28;
AUC0-t day28, truncated area under the plasma
concentration-time curve from the beginning of dosing to the last
measurable concentration on day 28; T1/2 day28,
half-life on day 28; T1/2 effective, half-life derived
from the observed antroquinonol accumulation; Ctrough
day14, trough plasma concentration on day 14; Ctrough
day27, trough plasma concentration on day 27; Ctrough
day28, trough plasma concentration on day 28.
Safety and efficacy assessments
The tumor burden was measured according to RECIST,
version 1.1, at baseline and at treatment discontinuation. Safety
evaluation included complete blood cell counts, biochemistry
laboratory data, C-reactive protein level, urinalysis, vital signs,
electrocardiogram (ECG) examination and adverse events (AEs).
Patients in the accelerated titration phase were
treated for 4 weeks at their first dose level, and then continued
with treatment in the extension program for 3 cycles if found to be
eligible. Moderate toxicity (MT) and DLTs were observed during the
initial 4-week treatment period. The accelerated phase ended when
either a DLT or MT was observed. A patient who experienced a DLT at
any other level besides the lowest level was returned to the
previous dose level. A patient who experienced a DLT at the lowest
dose level was discontinued from the study. Patients in the
standard titration phase were treated with antroquinonol for 4
weeks. DLTs were evaluated during the 4-week treatment period.
Prior to each 4-week cycle and at mid-cycle (i.e.,
day 14), a blood sample was drawn for complete blood testing in
order to determine whether it was safe to continue with the study
drug. If there was a ≥grade 3 toxicity, the patient's dose was
reduced to the next lower dose. Dose escalation was allowed in the
study and each patient was allowed two dose escalations. Patients
who required a dose reduction due to toxicity were not allowed dose
escalation at any point during the study. The patients were
observed for AEs until 6 weeks after their last treatment.
Extension program
The extension program included patients who had not
withdrawn from the study and wished to participate in the
accelerated or standard titration phase following their treatment
cycle. The patients were treated for up to three 4-week cycles for
as long as they were willing to continue, or until development of
intolerable toxicity or progressive disease.
Statistical analysis
The PKs variables were calculated from the serum
concentration data using standard, non-compartmental methods as
implemented in WinNonlin, version 5.2 or higher (Pharsight Corp.,
St. Louis, MO, USA). PKs parameters were presented with descriptive
statistics by dose level. Dose proportionality was to be evaluated
if sufficient data were available in dose groups.
All statistical analyses were performed using SAS
software, version 8.2 or higher (SAS Institute Inc. Cary, NC, USA).
Categorical data were summarized using counts and percentages. For
continuous variables, descriptive statistics were tabulated, such
as number of available observations, mean, median, standard
deviation, minimum and maximum. All available data and the
tabulation of the results were displayed by initial dose level and
with all levels pooled as a whole if applicable. All statistical
tests were two-sided, with a P-value of <0.05 indicating
statistically significant differences.
Results
Patients
A total of 13 patients (7 men and 6 women) were
enrolled in this study. All the patients were ethnic Chinese and
their mean age was 61 years (range, 43–86 years). Two patients
harboured EGFR-activating mutations, 4 patients had EGFR wild-type
and 7 patients had unknown EGFR status.
A total of 5 patients were enrolled in the
accelerated titration phase (1 patient each in the 50-, 100-, 200-,
300- and 450-mg dose groups) and 8 patients in the standard
titration phase (3 patients in the 450- and 5 patients in the
600-mg dose group). All 13 patients were included in the ITT and
the PK populations. The PP population consisted of 3 patients, with
1 patient in the 200- and 2 patients in the 600-mg dose group.
DLT and MTD
There were no DLTs reported in any patient for any
of the dose levels in the ITT population in the accelerated or
standard titration phase. An MTD was not determined in this study,
and was ≥600 mg daily.
Efficacy
Four patients continued to the fourth treatment
cycle, 1 patient each in the 200- and 450-mg dose groups and 2
patients in the 600-mg dose group. The best overall response was
stable disease in 3 patients (1 patient in the 200-mg dose group
and 2 patients in the 600-mg dose group), progressive disease in 7
patients, and not evaluated in 3 patients. Two patients were
identified with EGFR-activating mutations, including 1 patient in
the 450-mg dose group who discontinued the study drug prior to the
completion of the first cycle and 1 patient in the 600-mg dose
group who completed two cycles of treatment with the minor response
of the sum of the longest diameters of the target lesions being
reduced from 41 mm at screening to 31 mm at the end of the study
visits.
Safety
Overall, antroquinonol at all dose levels exhibited
a mild toxicity profile. A total of 4 patients reported 4 serious
AEs (2 patients each in the 450- and 600-mg dose groups, including
1 patient from each dose group who succumbed to progressive
disease). None of the mortalities, serious AEs or AEs leading to
discontinuation was related to the study drug. No grade 4
treatment-emergent AEs (TEAEs) were reported. Grade 3 TEAEs were
reported in 2 patients (1 patient in the 200-mg dose group
experienced vertigo and 1 patient in the 450-mg dose group
experienced decreased appetite). The most commonly occurring TEAEs
reported in >2 patients were diarrhea [10 patients, 76.9% (grade
1, 9 patients; and grade 2, 1 patient)], vomiting [9 patients,
69.2% (grade 1, 6 patients; and grade 2, 3 patients)], and nausea
[7 patients, 53.8% (grade 1, 6 patients; and grade 2, 1 patient)].
No patient exhibited any laboratory abnormalities recorded as
TEAEs, apart from 1 case of grade 1 hematuria in the 450-mg dose
group. There were mild increases or decreases in the hematological
and biochemical parameters, but none were considered as abnormal
results. None of the patients suffered from clinically significant
changes in any of the vital signs, ECG readings, or physical
examination parameters.
PK analysis
Due to the limited number of patients per dose group
(1 patient each received 50, 100, 200 and 300 mg, 4 patients
received 450 mg, and 5 patients received 600 mg), the PK results
should be interpreted with caution. The power model approach was
used to assess the dose proportionality of several antroquinonol
doses, ranging from 50 to 600 mg. The values for slope (95%
confidence interval) for area under the curve (AUC)0-t
and Cmax for day 1 were 0.641 (0.16–1.12) and 0.504
(0.17–0.83), respectively, and for day 28 they were 0.957
(0.59–1.32) and 0.682 (0.08–1.28), respectively. These results
indicate a dose-proportional increase over the dose range for
AUC0-t but not for Cmax under single-dose
conditions, and a dose-proportional increase over the dose range
for both AUC0-t and Cmax under multiple-dose
conditions.
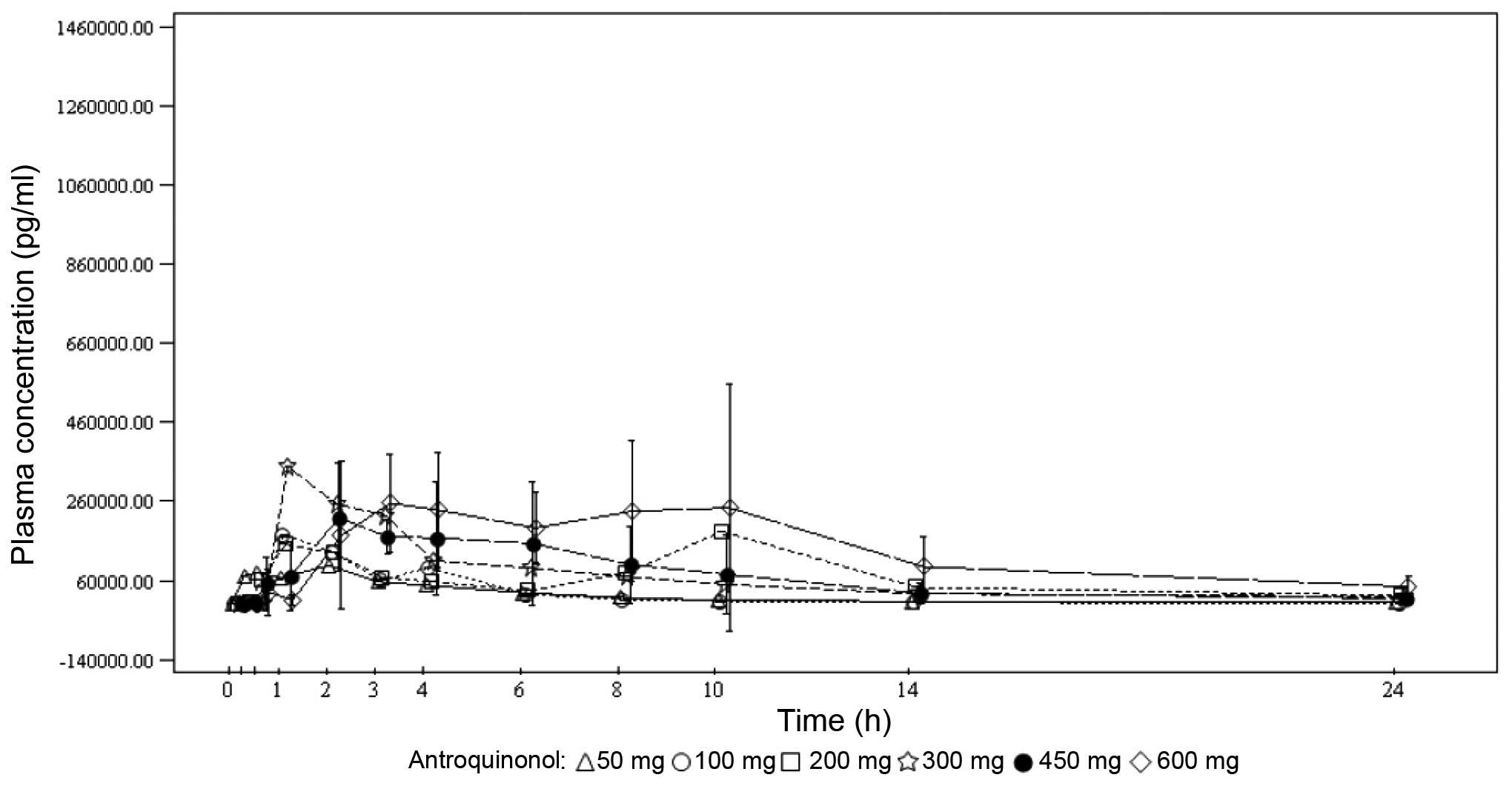
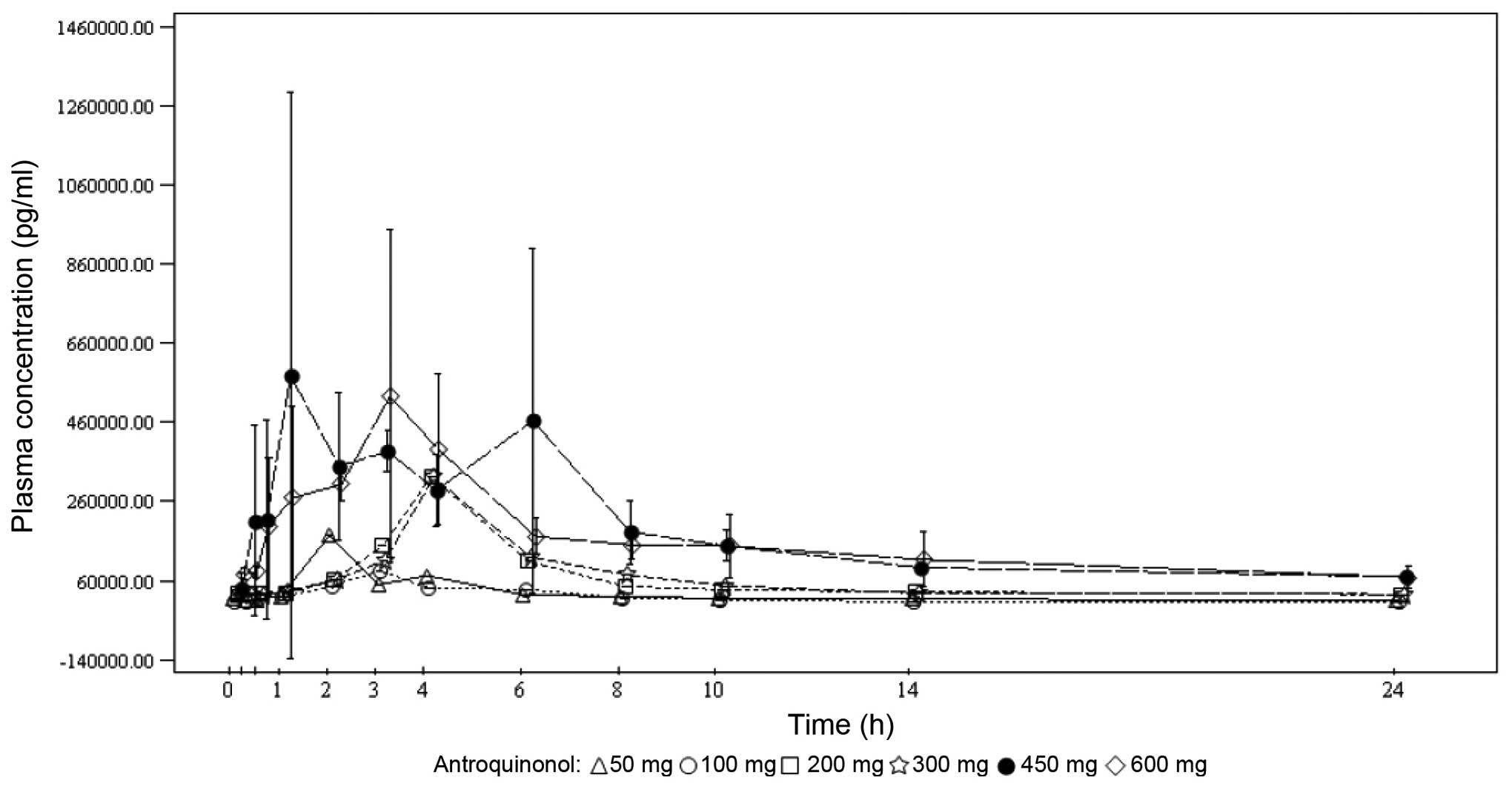
Under single-dose conditions, the maximum
antroquinonol concentration was generally observed between 1.00 and
3.70 h, with the exception of the 200-mg dose group, where a median
Tmax of 10.00 h was observed. Under multiple-dose
conditions, the results were similar with the median
Tmax ranging from 1.92 to 4.05 h. Antroquinonol was
rapidly eliminated following single and multiple administrations,
with a mean T1/2 ranging from 1.30 to 4.33 h,
independently of the treatment dose. The trough plasma
concentrations were similar on days 14, 27 and 28 within each dose
group, and increased with the treatment dose. These results suggest
that the steady state was probably attained on day 14. In terms of
accumulation, no clear trend was observed when comparing the rate
and extent of absorption on day 1 to those on day 28. The PKs mean
plasma concentrations vs. time by treatment dose on days 1 and 28
are presented in Figs. 2 and 3, respectively.
Discussion
Antrodia camphorata is an endemic species in
Taiwan and has been traditionally used for food and drug
intoxication, and for the treatment of diarrhea, abdominal pain,
hypertension, pruritus and liver cancer (7). Other studies have reported Antrodia
camphorata to possess detoxifying, anti-inflammatory,
immunomodulatory, hepatoprotective and anticancer properties
(8–11).
Extracts of Antrodia camphorata have been
used in China as an alternative anticancer agent for decades.
Previous in vitro studies have demonstrated that
antroquinonol displays anticancer activity against several cancer
cell lines, including the A549 pulmonary adenocarcinoma cell line
(3–5).
The present first-in-human study further investigated the DLT and
MTD of antroquinonol and its recommended dose for phase II studies
in different types of cancer.
Antroquinonol at all dose levels, administered daily
for 4 weeks, was generally found to be safe and tolerable. The AEs
were mainly gastrointestinal, including diarrhea, vomiting and
nausea. Since no DLTs occurred in this study, the MTD could not be
determined. The overall response was stable disease in 3 patients
and progressive disease in 7 patients. Among the 5 patients in the
600-mg dose level, 3 patients were evaluable for treatment response
and 2 achieved stable disease for >3 months. However, these data
should be interpreted with caution due to the limited number of
patients in this study.
It may have been preferable to perform tissue biopsy
and/or serum biomarker examination during antroquinonol treatment
to document its effects on signal transduction pathways or tumor
cell cycles when the patients were under different dose levels.
However, this part was omitted, as only few patients with different
dose levels of treatment were expected to be evaluable and it was
difficult to persuade patients to undergo another tissue biopsy or
further blood tests for biomarker studies. In addition, biomarker
studies, including tissue sample studies that will be performed in
the phase II study, would be more appropriate, as higher patient
numbers and more biomarker findings may be correlated with
treatment efficacy.
In conclusion, antroquinonol administered up to a
dose level of 600 mg daily for 4 weeks was generally safe and
tolerable, as no particular safety concerns or DLTs were identified
in the present study. Progression of antroquinonol to further
clinical development is required. The data generated from the
safety and PK profiles in this study support further studies of
antroquinonol in a larger number of cancer patients. The
recommended phase II dose is at least 600 mg daily, since DLTs did
not occur in the present study.
Acknowledgements
The authors would like to thank the participating
patients and their families, as well as Dr Howard Cheng (Clinical
Research R&D, Golden Biotechnology Corp., New Taipei, Taiwan)
for his contribution to this study.
References
|
1
|
Pfister DG, Johnson DH, Azzoli CG, et al:
American Society of Clinical Oncology treatment of unresectable
non-small-cell lung cancer guideline: update 2003. J Clin Oncol.
22:330–353. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chiou JF, Wu AT, Wang WT, Kuo TH, Gelovani
JG, Lin IH, Wu CH, Chiu WT and Deng WP: A preclinical evaluation of
Antrodia camphorata alcohol extracts in the treatment of
non-small cell lung cancer using non-invasive molecular imaging.
Evid Based Complement Alternat Med. 2011:9145612011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chiang PC, Lin SC, Pan SL, Kuo CH, Tsai
IL, Kuo MT, Wen WC, Chen P and Guh JH: Antroquinonol displays
anticancer potential against human hepatocellular carcinoma cells:
a crucial role of AMPK and mTOR pathways. Biochem Pharmacol.
79:162–171. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yu CC, Chiang PC, Lu PH, Kuo MT, Wen WC,
Chen P and Guh JH: Antroquinonol, a natural ubiquinone derivative,
induces a cross talk between apoptosis, autophagy and senescence in
human pancreatic carcinoma cells. J Nutrition Biochem. 23:900–907.
2012. View Article : Google Scholar
|
|
5
|
Kumar VB, Yuan TC, Liou JW, Yang CJ, Sung
PJ and Weng CF: Antroquinonol inhibits NSCLC proliferation by
altering PI3K/mTOR proteins and miRNA expression profiles. Mutation
Res. 707:42–52. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Eisenhauer EA, Therasse P, Bogaerts J, et
al: New response evaluation criteria in solid tumours: revised
RECIST guideline (version 1.1). Eur J Cancer. 45:228–247. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen CC, Shiao YJ, Lin RD, Shao YY, Lai
MN, Lin CC, Ng LT and Kuo YH: Neuroprotective diterpenes from the
fruiting body of Antrodia camphorata. J Nat Prod.
69:689–691. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee TH, Lee CK, Tsou WL, Liu SY, Kuo MT
and Wen WC: A new cytotoxic agent from solid-state fermented
mycelium of Antrodia camphorata. Planta Med. 73:1412–1415.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hseu YC, Chang WC, Hseu YT, Lee CY, Yech
YJ, Chen PC and Yang HL: Protection of oxidative damage by aqueous
extract from Antrodia camphorata mycelia in normal human
erythrocytes. Life Sci. 71:469–482. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chang JM, Lee YR, Hung LM, Liu SY, Kuo MT,
Wen WC and Chen P: An extract of Antrodia camphorata mycelia
attenuates the progression of nephritis in systemic lupus
erythematosus-prone NZB/W F1 mice. Evid Based Complement Alternat
Med. 2011:4658942011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Song TY and Yen GC: Protective effects of
fermented filtrate from Antrodia camphorata in submerged
culture against CC14-induced hepatic toxicity in rats. J Agric Food
Chem. 51:1571–1577. 2003. View Article : Google Scholar : PubMed/NCBI
|