Introduction
Acute lymphoblastic leukemia (ALL) is a common type
of clonal hematopoietic malignant disease of lymphocyte precursor
cells (1). Clinical manifestations,
such as anemia, bleeding, infection, as well as enlargement of the
liver, spleen and lymph nodes, may occur due to the infiltration of
tissues and organs throughout the body by leukemic cells. Bone
marrow necrosis (BMN) and acute renal insufficiency (ARI) are rare
and frequently fatal conditions, which may occur as a primary or
secondary manifestation of ALL (2,3).
However, it is extremely rare for BMN and ARI to occur
simultaneously in a patient with Philadelphia chromosome-positive
(Ph+) ALL and, to the best of our knowledge, such a case
has not been reported to date. It is documented that Ph+
ALL is associated with at least a 10% reduction of primary complete
remission by standard induction chemotherapy, and long-term
prognosis remains poor compared with that of Ph− ALL
(4).
We herein describe the case of a patient with
Ph+ ALL concomitant with BMN and ARI, who was treated
with imatinib combination and maintenance therapy, allogeneic
hematopoietic stem-cell transplantation (allo-HSCT), and various
comprehensive treatments, such as repeated hemodialysis,
anti-infective therapy and transfusion of platelets and erythrocyte
suspension.
Case report
A 23-year-old male patient with paroxysmal bone pain
and intermittent pyrexia for a duration of 2 months was referred to
the Department of Hematology of the Affiliated Hospital of Xuzhou
Medical University during April, 2015. Upon admission, the results
of the emergency blood routine tests were as follows: White blood
cell (WBC) count 41.8×109/l, hemoglobin concentration
103 g/l and platelet count 104×109/l; the blast
proportion was 21%. The results of the blood biochemical tests were
as follows: Serum creatinine (SCr) 0.95 mg/dl, blood urea nitrogen
(BUN) 19.97 mg/dl, uric acid 7.77 mg/dl, calcium 9.58 mg/dl,
lactate dehydrogenase (LDH) 2263 U/l and alkaline phosphatase (ALP)
214 U/l.
Following admission, the patient's condition
deteriorated rapidly, with recurrent hyperpyrexia (40.2°C), chills,
nausea and vomiting. The patient also had severe systemic
arthralgia and oliguria (~200 ml/24 h). Upon examination,
tachypnea, tachycardia, ecchymoses and pitting oedema of the lower
extremities were observed. Blood routine tests revealed a
significant decline in the WBC count (from 41.8 to
11.7×109/l), hemoglobin level (95 g/l) and platelet
count (2×109/l). The results of the biochemical
laboratory test were as follows: SCr 5.67 mg/dl, BUN 86.35 mg/dl,
uric acid 18.31 mg/dl, calcium 6.17 mg/dl, phosphorus 10.31 mg/dl,
and an LDH level increased up to 3,778 U/l. Abdominopelvic
ultrasonography revealed normal-sized kidneys, with increased
parenchymal echogenicity and absence of hydronephrosis, and a void
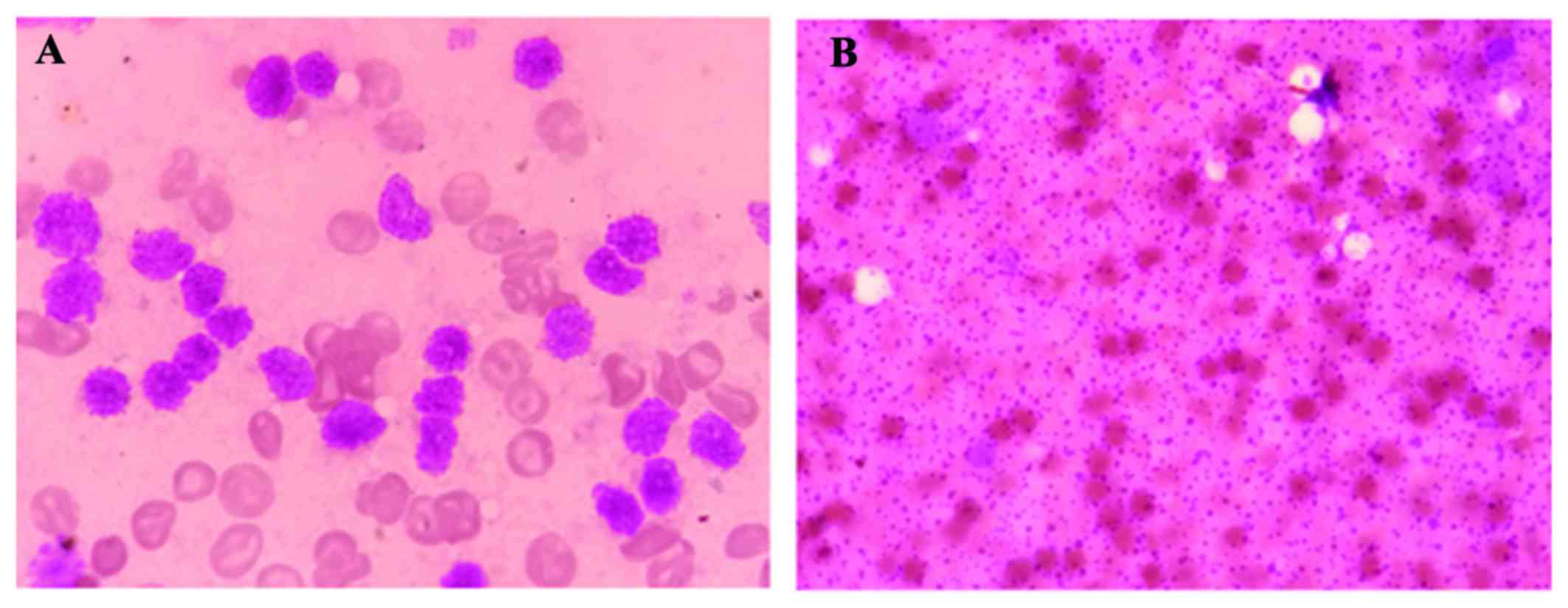
bladder. For diagnosis, BM aspirate samples were obtained, which
were brown in colour, and the cell morphology indicated BMN
(Fig. 1A and B). A BM biopsy
specimen revealed hypercellularity in lymphocytes, with 20%
lymphoblasts. Flow cytometric immunophenotyping of BM cells
revealed that 96.9% were CD19-positive, 85.9% were CD10-positive,
47.2% were CD10/CD33-positive, 34.8% were CD20-positive, 90.6% were
CD34/HLA-DR-positive, 2.2% were cCD79a-positive, and 46.2% were
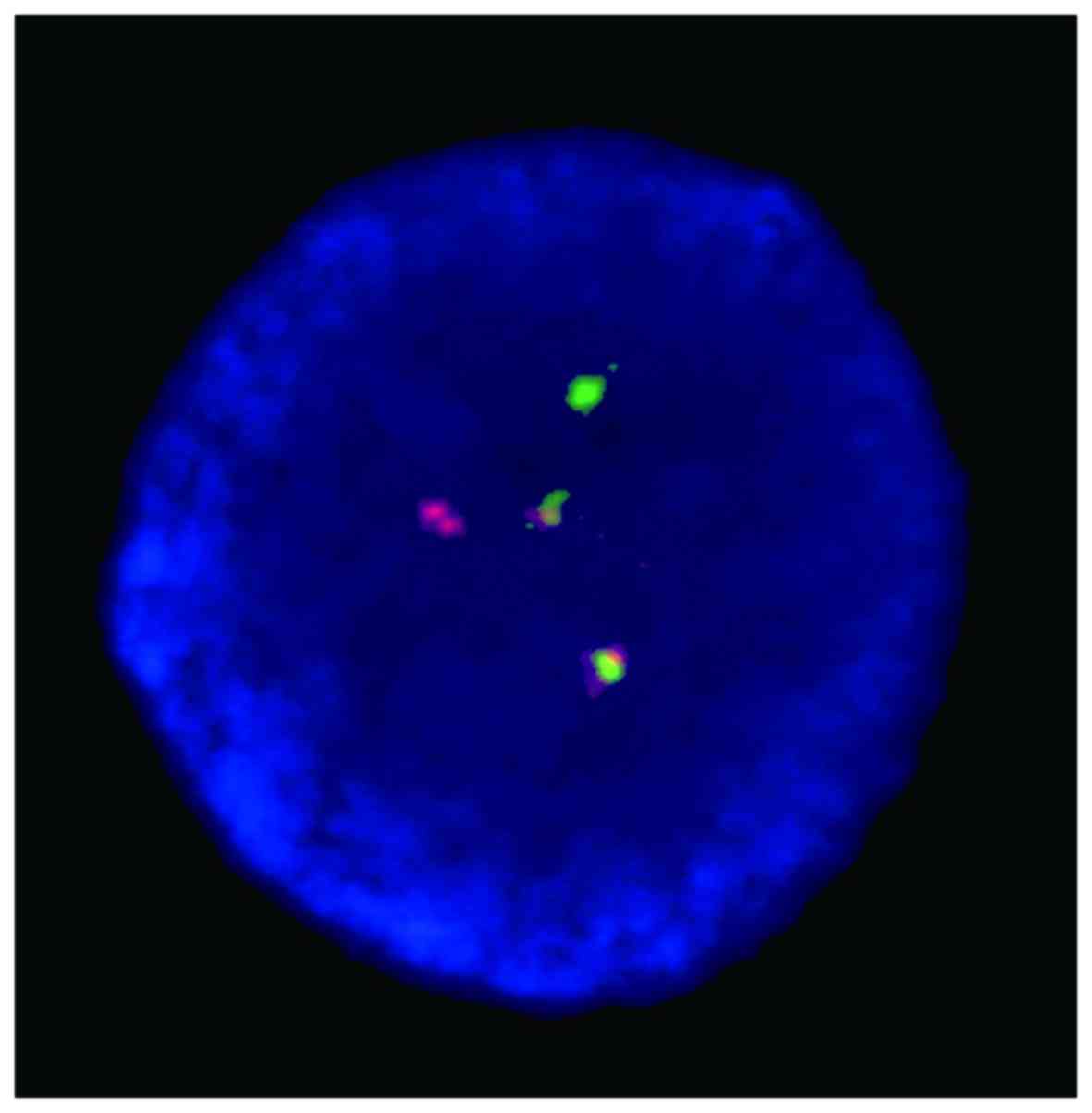
cCD22-positive. Fluorescence in situ hybridization (FISH)
came back positive for the BCR-ABL fusion gene, and the BCR-ABL/ABL
ratio was 82.82% (Fig. 2).
Therefore, the patient was diagnosed with Ph+ ALL
(B-cell) complicated with BMN and ARI based on the medical history,
clinical manifestations and laboratory test results.
Subsequent to admission, treatment was initiated,
including antipyretic therapy, rehydration, energy replenishment
[daily treatment with cytidine disodium triphosphate (80 mg),
inosine (400 mg), 50% glucose (40 g), ascorbic acid (1 g) and
insulin (16 units) for a total of 7 days] and oral tramadol for
analgesia. On the third day after admission, prednisolone (600
mg/day), allopurinol and furosemide were administered, along with
transfusion of platelets and erythrocyte suspension, hemodialysis
with therapy for acid-base imbalance, electrolyte disorders, and
anti-infection with imipenem. At 10 days after treatment
initiation, the patient's clinical status had significantly
improved, urine output gradually increased, the oedema of the lower
extremities subsided, and the levels of SCr, BUN and uric acid had
almost returned to normal; therefore, dialysis was terminated.
Thereafter, the patient received cyclophosphamide (CY; 600 mg/day)
to reduce the tumor load, and prednisolone administration was
continued at 400 mg/day. On the 12th day after admission, the
patient received imatinib combined with induction chemotherapy
(imatinib plus VIP), with imatinib administered in combination with
vindesine (4 mg/day) on days 1, 8, 15 and 22; idarubicin (10
mg/day) was administered for the first 3 consecutive days, with
imatinib (400 mg/day) and prednisolone (60 mg/day) administered
daily. In addition, the patient continually received supportive
treatment, including hydration, alkalization, hepatoprotection and
renoprotection, energy replenishment as previously aforementioned
and anti-infection treatment. On day 14 of chemotherapy, the VIP
regimen was discontinued due to severe BM suppression, while
transfusion of erythrocytes and platelets was performed, along with
administration of recombinant granulocyte colony-stimulating factor
to increase the WBC count. On the 42nd day of hospitalization, the
results of routine blood tests had nearly returned to normal, a BM
smear revealed no signs of necrosis and null lymphoblasts,
indicating partial remission of the primary disease. Thereafter,
the patient received the second course of chemotherapy (imatinib
plus VDCLP), with imatinib (400 mg/day) administered daily in
combination with vindesine (4 mg/day) on days 1, 8, 15 and 22;
daunorubicin (40 mg/day) was administered on days 1–3, CY (1.0
g/day) on days 1 and 15, L-asparaginase (10,000 U) on alternate
days from day 10 to 22 and prednisolone (60 mg/day) from day 1 to
28. Comprehensive treatments were also performed as described
above, and the prednisolone dose was reduced gradually from day 15
onwards and discontinued on day 28. On the 72nd day of hospital
stay, the results of routine blood tests were normal for the three
types of blood cells, BM examination revealed 0% lymphoblasts, and
cytogenetic examination of BM cells revealed a normal karyotype and
null BCR/ABL, indicating that hematological remission as well as
complete remission at the genetic and molecular level had been
achieved. Subsequently, single-agent imatinib was administered as
maintenance chemotherapy at 400 mg/day. Three weeks later, an
intensified chemotherapy with high-dose methotrexate and
vincristine + prednisone was administered, with methotrexate 3 g
and vincristine 30 mg on day 1 and prednisone (30 mg/day) on days
1–7. After the third course of chemotherapy, pretreatment
chemotherapy with the improved busulfan (BU)/CY regimen (Ara-C 5.0
g on days −10 and −9, BU 48 mg q6 h × 3 d on days-8 to −6, CY 28 mg
on days −5 and −4, lomustine 450 mg and anti-thymocyte globulin 240
mg on days −5 to −2) was performed. During August 2015, the patient
underwent allo-HSCT. One month after allo-HSCT, rapid and complete
hematopoietic reconstitution was achieved, and the patient showed
no major complications occurred. Repeated FISH tests revealed
negative expression of the BCR-ABL fusion gene. A total of 5 months
after allo-HSCT, treatment with imatinib (300 mg/day) was resumed
in order to prevent relapse. Consider that leukemia can impair the
central nervous system and periocular tissues, the patient was
pretreated with the fludarabine and cyclophosphamide regimen
[fludarabine phosphate (50 mg; once daily on days 1–3); and
cyclophosphamide (1.2 g; once daily on days 1 and 2)] between April
6 and 8, 2017. Following this, chimeric antigen receptor (CAR)-T
cells present in peripheral blood obtained from the sibling of the
patient (1.73×106 /kg) were intravenously infused on
April 11, 2017. During May 2017, the patient was discharged from
hospital and was in remission. The final follow-up appointment was
performed 2 years following initial diagnosis, and the patient
remained in remission.
Discussion
BMN is characterized by coagulation, architectural
destruction and necrosis of a large area of hematopoietic and
stromal tissues within the BM (5,6). In
addition, high levels of LDH and ALP are common laboratory findings
in the majority of cases with BMN (5,7). Rather
than the typical signs or symptoms of leukemia in the early stages,
our patient presented with pyrexia and arthralgia, a progressive
decrease in blood cell counts, and elevated LDH and ALP levels, all
of which strongly suggested a diagnosis of BMN. Consequently, based
on a series of tests, a diagnosis of Ph+ ALL (B-cell)
complicated with BMN was confirmed.
The mechanism underlying ALL as a contributor to BMN
has not been fully elucidated. Routine blood tests upon hospital
admission revealed elevated WBC count of 41.8×109/l, and
cytological examination of BM revealed elevation of the lymphocytic
proportion (20% lymphoblasts). Thus, BMN may be attributable to the
microcirculatory disturbance due to an overload of lymphoblasts
(8). The patient presented with
symptoms of tumor necrosis factor (TNF)-α toxicity at early onset
manifesting as hyperpyrexia, chills, headache, nausea and fatigue.
Therefore, TNF-α may play a pivotal role in the pathogenesis of BMN
(7,9). Since the patient received no special
medication prior to the diagnosis of BMN, the possibility of BMN
caused by drugs or poisons may be excluded.
ARI is defined as a sudden reduction in renal
function, characterized by an absolute increase in serum SCr level
to ≥0.3 mg/dl or by 50% compared with baseline (10). ARI is a common and serious
complication associated with malignancy, which occurs in various
clinical settings for numerous reasons (11–13). In
the present case, at 32 h after admission, the patient experienced
nausea, vomiting, oliguria and oedema in the bilateral lower
extremities. Renal function test results revealed a SCr level of
5.67 mg/dl (>1.5-fold from baseline, with an absolute increase
of 4.48 mg/dl, >0.3 mg/dl), BUN 86.35 mg/dl and uric acid 18.31
mg/dl prior to the induction of chemotherapy, indicating the
development of ARI according to the relevant diagnostic criteria
(10).
Despite the fact that the mechanisms through which
ALL contributes to ARI are not clearly understood, ARI may be an
important sequela of the tumor lysis syndrome (TLS). TLS is a term
used to describe a series of metabolic abnormalities and multiple
organ dysfunctions that result from the rapid release of the
intracellular contents of lysed tumor cells. TLS is classified into
laboratory and clinical TLS, with the former involving at least two
abnormalities in serum uric acid, potassium, phosphorus or calcium
levels (14). Clinical TLS is
defined as the presence of laboratory TLS and one of the following:
ARI, cardiac arrhythmias, or seizures. It has been reported that
patients with ALL or non-Hodgkin lymphoma have a high incidence of
TLS, and that TLS may be the leading cause of ARI in such patients
(15,16). Additional univariate analysis
revealed that TLS develops more frequently in patients with
elevated serum uric acid, SCr, LDH and WBC count (17,18). In
the present case, the patient fit the criteria of TLS with respect
to 3 items of laboratory results (hyperuricemia >8.0 mg/dl,
hyperphosphatemia >4.6 mg/dl and hypocalcemia <7.0 mg/dl) and
one clinical symptom (ARI), accompanied by high-risk
characteristics, namely increased LDH level and WBC count;
therefore, our patient presented with clinical TLS (14).
The pathogenesis of ARI resulting from TLS is
complex and multifactorial. Of the numerous factors that can cause
ARI, uric acid and phosphorus are the most common. Hyperuricemia is
the main characteristic of TLS metabolic disturbance, which results
from the breakdown of purine-containing nucleic acids due to lysed
tumor cells. Previous studies demonstrated that hyperuricemia plays
a major role in the pathophysiological process of ARI associated
with TLS (19,20). In addition, new evidence suggests
that uric acid may also cause ARI through a
crystallopathy-independent mechanism observed in TLS, such as renal
vasoconstriction, renal ischemia, oxidation and inflammation
(21). Similarly, as a result of
massive release of intracellular phosphate stores,
hyperphosphatemia is also characteristic of TLS, and the
accumulation of calcium phosphate in the renal tubules may also be
involved in the pathogenesis of ARI in patients with TLS (22). Our patient exhibited a persistent
elevation of blood uric acid and phosphorus levels and decreased
calcium levels. Therefore, hyperuricemia and hyperphosphatemia may
account for TLS-induced ARI in this patient. A case of leukemic
infiltration into the kidneys presenting as ARI has been reported
(23,24). Unfortunately, our patient refused to
undergo renal biopsy; thus, it remains uncertain whether leukemic
cell infiltration was associated with ARI development.
Intriguingly, despite the low complete remission
rates and shortened remission and disease-free survival duration in
Ph+ ALL patients by conventional chemotherapy, a recent
study demonstrated that imatinib can improve the complete remission
rate at the early stage, prolong disease-free survival, and improve
the prognosis of Ph+ ALL patients (25). The present case further supports that
imatinib may be a key treatment component for patients with
Ph+ ALL with potentially fatal complications, which may
improve the conditions for subsequent allo-HSCT, even if this
assertion has yet to be supported by more studies.
In summary, the main conclusions of this case study
are as follows: First, in a patient with BMN and/or ARI, a
diagnosis of ALL should be considered; and second, Ph+
ALL complicated by BMN and ARI is a rare and serious condition, and
timely accurate diagnosis and effective management are crucial for
successful treatment and for improving the quality of life of such
patients.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
Not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
The patient provided written informed consent for
the publication of clinical data.
Authors' contributions
JX and KX were responsible for the data analysis and
interpretation. HS, ZY, KZ and FZ collected scientific literature
and clinical information. JX and FZ wrote and edited the
manuscript. All authors have read and approved of the final version
of this manuscript.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Davi F, Gocke C, Smith S and Sklar J:
Lymphocytic progenitor cell origin and clonal evolution of human
B-lineage acute lymphoblastic leukemia. Blood. 88:609–621.
1996.PubMed/NCBI
|
|
2
|
Montemayor-Montoya JL, De León-Cantú RE,
Gómez-Almaguer D and Herrera-Garza JL: 3 cases of bone marrow
necrosis in acute leukemia. Rev Invest Clin. 49:295–298. 1997.(In
Spanish). PubMed/NCBI
|
|
3
|
Canet E, Zafrani L, Lambert J, Thieblemont
C, Galicier L, Schnell D, Raffoux E, Lengline E, Chevret S, Darmon
M and Azoulay E: Acute kidney injury in patients with newly
diagnosed high-grade hematological malignancies: Impact on
remission and survival. PLoS One. 8:e558702013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moorman AV, Harrison CJ, Buck GA, Richards
SM, Secker-Walker LM, Martineau M, Vance GH, Cherry AM, Higgins RR,
Fielding AK, et al: Karyotype is an independent prognostic factor
in adult acute lymphoblastic leukemia (ALL): Analysis of
cytogenetic data from patients treated on the Medical Research
Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG)
2993 trial. Blood. 109:3189–3197. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bernard C, Sick H, Boilletot A and
Oberling F: Bone marrow necrosis. acute microcirculation failure in
myelomonocytic leukemia. Arch Intern Med. 138:1567–1569. 1978.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Paydas S, Ergin M, Baslamisli F, Yavuz S,
Zorludemir S, Sahin B and Bolat FA: Bone marrow necrosis:
Clinicopathologic analysis of 20 cases and review of the
literature. Am J Hematol. 70:300–305. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lackner H, Strenger V, Sovinz P,
Beham-Schmid C, Pilhatsch A, Benesch M, Schwinger W, Ulreich R,
Schmidt S and Urban C: Bone marrow necrosis in a girl with
Hodgkin's disease. Support Care Cancer. 20:2231–2234. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Moritake H, Obara M, Sameshima N, Asada Y,
Komatsu H, Hyakuna N, Sugita K, Ishida Y, Kato M, Tanizawa A, et
al: Analysis of the molecular mechanism underlying bone marrow
necrosis with acute lymphoblastic leukemia. Int J Hematol.
102:349–356. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Seki Y, Koike T, Yano M, Aoki S, Hiratsuka
M, Fuse I and Aizawa Y: Bone marrow necrosis with dyspnea in a
patient with malignant lymphoma and plasma levels of
thrombomodulin, tumor necrosis factor-alpha, and D-dimer. Am J
Hematol. 70:250–253. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kellum JA, Lameire N, Aspelin P, Barsoum
RS, Burdmann EA, Goldstein SL, Herzog CA, Joannidis M, Kribben A,
Levey AS, et al: Kidney Disease: Improving Global Outcomes (KDIGO)
Acute Kidney Injury Work Group: KDIGO clinical practice guideline
for acute kidney injury. Kidney Int Suppl. 2:1–138. 2012.
|
|
11
|
Abu-Alfa AK and Younes A: Tumor lysis
syndrome and acute kidney injury: Evaluation, prevention, and
management. Am J Kidney Dis. 55 5 Suppl 3:S1–S13; quiz S14-S19.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Soares M, Salluh JI, Carvalho MS, Darmon
M, Rocco JR and Spector N: Prognosis of critically ill patients
with cancer and acute renal dysfunction. J Clin Oncol.
24:4003–4010. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Canet E, Vincent F, Darmon M and Soares M:
Acute kidney injury in hematological patients. Curr Opin Crit Care.
21:549–558. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cairo MS and Bishop M: Tumour lysis
syndrome: New therapeutic strategies and classification. Br J
Haematol. 127:3–11. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stapleton FB, Strother DR, Roy S III,
Wyatt RJ, McKay CP and Murphy SB: Acute renal failure at onset of
therapy for advanced stage Burkitt lymphoma and B cell acute
lymphoblastic lymphoma. Pediatrics. 82:863–869. 1988.PubMed/NCBI
|
|
16
|
Annemans L, Moeremans K, Lamotte M, Conde
Garcia J, van den Berg H, Myint H, Pieters R and Uyttebroeck A:
Incidence, medical resource utilisation and costs of hyperuricemia
and tumour lysis syndrome in patients with acute leukaemia and
non-Hodgkin's lymphoma in four European countries. Leuk Lymphoma.
44:77–83. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Coiffier B, Altman A, Pui CH, Younes A and
Cairo MS: Guidelines for the management of pediatric and adult
tumor lysis syndrome: An evidence-based review. J Clin Oncol.
26:2767–2778. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oiwa K, Morita M, Kishi S, Okura M, Tasaki
T, Matsuda Y, Tai K, Hosono N, Ueda T and Yamauchi T: High risk of
tumor lysis syndrome in symptomatic patients with multiple myeloma
with renal dysfunction treated with bortezomib. Anticancer Res.
36:6655–6662. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kjellstrand CM, Cambell DC II, von
Hartitzsch B and Buselmeier TJ: Hyperuricemic acute renal failure.
Arch Intern Med. 133:349–359. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tsokos GC, Balow JE, Spiegel RJ and
Magrath IT: Renal and metabolic complications of undifferentiated
and lymphoblastic lymphomas. Medicine (Baltimore). 60:218–229.
1981. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shimada M, Johnson RJ, May WS Jr,
Lingegowda V, Sood P, Nakagawa T, Van QC, Dass B and Ejaz AA: A
novel role for uric acid in acute kidney injury associated with
tumour lysis syndrome. Nephrol Dial Transplant. 24:2960–2964. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Prasada H: Sevelamer hydrochloride for
tumor lysis syndrome-related hyperphosphatemia. Indian Pediatr.
52:613–615. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Uprety D, Peterson A and Shah BK: Renal
failure secondary to leukemic infiltration of kidneys in CLL-a case
report and review of literature. Ann Hematol. 92:271–273. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Spapen J, Fostier K, De Raeve H, Janssens
P and Spapen H: An unexpected complication of chronic
myelomonocytic leukemia: Severe renal failure due to malignant
tubulo-interstitial cell infiltration. Int J Nephrol Renovasc Dis.
9:1–4. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lim SN, Joo YD, Lee KH, Kim DY, Lee JH,
Lee JH, Chi HS, Yun SC, Lee WS, Lee SM, et al: Long-term follow-up
of imatinib plus combination chemotherapy in patients with newly
diagnosed Philadelphia chromosome-positive acute lymphoblastic
leukemia. Am J Hematol. 90:1013–1020. 2015. View Article : Google Scholar : PubMed/NCBI
|