Introduction
Glioma accounts for ~80% of primary malignant brain
tumors in adults, and progresses rapidly and results in increased
rates of mortality compared with any other type of tumor (1). Temozolomide (TMZ), an oral alkylating
agent, is the first line chemotherapeutic drug in current standard
glioma treatment (2).
The therapeutic benefit of TMZ depends on its
ability to alkylate DNA, which often occurs at the N-7 or O-6
position of guanine residues. The minor adduct
O6-methylguanine (O6MeG) is the most
cytotoxic lesion, which mismatches with thymine. The resulting
O6MeG/T mismatches are recognized by the mismatch repair
system, which performs futile repair cycles and results in DNA
double-strand breaks (DSBs) (3,4).
Following DNA damage, ataxia-telangiectasia mutated (ATM), a
serine/threonine protein kinase, is recruited to DNA foci, which in
turn activates ATM. Activated ATM then transmits the DNA damage
signal to downstream substrates and elicits DNA damage responses
(5–7).
The emergence of drug resistance often leads to
therapeutic failure in the treatment of glioma, precluding
long-term survival of the patients. The primary cytotoxic lesion,
the O6MeG DNA adduct, may be eliminated by
O6-methylguanine DNA methyltransferase (MGMT) in gliomas
expressing this DNA repair enzyme (4,8).
Since the MGMT promoter in almost half of the glioblastoma
specimens was methylated and the MGMT promoter methylation status
of the primary tumor was retained at recurrence, other
chemoresistance mechanisms are critical for TMZ tolerance in
MGMT-negative glioblastoma (9,10).
Autophagy, which is characterized by the formation
of acidic vesicular organelles (AVOs), is another cellular process
critical for glioma cell survival under TMZ treatment. Induction of
autophagy by TMZ has been documented in glioma cell lines and
surgical specimens, and inhibition of autophagy augments
TMZ-induced apoptosis in glioma cells (11,12).
However, the molecular mechanism by which TMZ induces autophagy is
largely unknown.
In the present study, it was hypothesized that
TMZ-induced activation of ATM elicited autophagy. In order to
assess this hypothesis, the effect of ATM inhibition on autophagy
was evaluated, the activation of AMPK and ULK1 was assessed, which
were autophagy-initiating kinases, under ATM inhibition, and their
association was investigated.
Materials and methods
Cell culture and reagents
U87MG and U251 human malignant glioma cell lines
were obtained from the Cell Bank of the Chinese Academy of Sciences
(Shanghai, China). Cells were maintained in Dulbecco’s modified
Eagle’s medium (Gibco-BRL, Carslbad, CA, USA) supplemented with 10%
fetal bovine serum (PAA Laboratories, Pasching, Australia) at 37°C
in a 5% CO2-humidified atmosphere. TMZ was supplied by
Tasly Pharmaceutical Co., Ltd. (Tianjin, China). ATM kinase
inhibitor KU-55933 (sc-202963) and the AMPK inhibitor compound C
(P5499) were purchased from Santa Cruz Biotechnology, Inc. (Santa
Cruz, CA, USA) and Sigma-Aldrich (St. Louis, MO, USA),
respectively. TMZ, KU-55933 and compound C were dissolved in
dimethylsulfoxide (DMSO; Sigma-Aldrich). The final concentration of
DMSO in the culture medium did not exceed 0.01%, thus did not
effect cell viability or protein expression.
Protein extracts
For experiments using whole-cell lysate samples,
cells were washed with ice-cold phosphate-buffered saline (PBS),
removed from the culture dishes and incubated in lysis buffer (150
mM NaCl, 0.1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM
Tris, 1 mM DTT, 5 mM Na3VO4, 1 mM
phenylmethylsulfonyl fluoride, 10 μg/ml trypsin inhibitor, 10 μg/ml
aprotinin and 5 μg/ml leupeptin, pH 7.4) for 30 min on ice. The
lysate was centrifuged at 12,000 × g for 30 min at 4°C and the
supernatant was collected as a whole cell lysate. For experiments
using subcellular fractionation, cells were washed with ice-cold
PBS, removed from the culture dishes and incubated in hypotonic
protein extract buffer (1 mM EGTA, 1 mM EDTA, 10 mM HEPES, 10 mM
KCl, 10 mM NaF, 1 mM Na3VO4, 1 mM
dithiothreitol, 10 mM β-glycerophosphate, 100 mg/ml
phenylmethylsulfonyl fluoride and 10 mg/ml aprotinin) for 10 min on
ice, and lysed by the addition of Igepal CA-630 (final
concentration, 0.4%) with vigorous mixing for 10 sec. The lysate
was centrifuged at 12,000 × g for 5 min at 4°C and the supernatant
was collected as a cytoplasmic protein extract. The pellet was
sonicated using an Ultrasonic homogenizer (BL-96-II L; Voshin
Instrument Co., Wuxi, Jiangsu, China) and incubated in hypertonic
protein extract buffer (10 mM Tris, 1 mM EGTA, 1 mM EDTA, 400 mM
NaCl, 10 mM NaF, 1 mM Na3VO4, 1 mM
dithiothreitol, 10 mM β-glycerophosphate, 0.5% Igepal CA-630, 100
mg/ml phenylmethylsulfonyl fluoride and 10 mg/ml aprotinin) for 30
min on ice and centrifuged at 12,000 × g for 5 min, and the
supernatant was collected as a nuclear protein extract. All protein
samples were stored at −80°C.
Western blot analysis
Western blot analysis was performed using standard
methods. β-actin (4970, Cell Signaling Technology, Inc., Danvers,
MA, USA) was used as a loading control. The other primary
antibodies used were as follows: Monoclonal anti-phospho-ATM
(Ser1981) antibody (5883, Cell Signaling Technology Inc., Danvers,
MA, USA), monoclonal anti-ATM antibody (2873, Cell Signaling
Technology Inc.), polyclonal anti-AMPKα antibody (2532, Cell
Signaling Technology Inc.), monoclonal anti-phospho-AMPKα (Thr172)
antibody (2535, Cell Signaling Technology Inc.), polyclonal
anti-phosphor-ULK1 (Ser467) antibody (4634, Cell Signaling
Technology Inc.), polyclonal anti-LC3B antibody (2775, Cell
Signaling), polyclonal anti-γH2AX antibody (ab2893, Abcam,
Cambridge, MA, USA). Microtubule-associated protein light chain 3B
(LC3B), which is cleaved into LC3B-I and LC3B-II during autophagy,
was used as an autophagy marker (13–15).
Detection of AVOs
Quantification of autophagy by acridine orange
staining using flow cytometry was performed as described previously
(16,17). Briefly, following drug treatment,
acridine orange (sc-358795, Santa Cruz Biotechnology, Inc.) was
added at a final concentration of 1 μg/ml for a period of 15 min.
All floating and adherent cells were collected, washed with PBS,
resuspended with phenol red-free growth medium and analyzed by flow
cytofluorometry (guava easyCyte 5HT; Millipore Corporation,
Hayward, CA, USA). When excited with a 488-nm laser, the nucleolus
of acridine orange-stained cells fluoresced bright green and acidic
vesicles emitted bright red fluorescence. The forward scatter
threshold was adjusted to omit cellular debris and 5,000 ungated
events were analyzed. Cells containing AVOs were identified as
double positive cells.
Detection of apoptosis
Cell apoptosis was detected with an Annexin
V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis
detection kit (C1063, Beyotime Biotech., Jiangsu, China) according
to the manufacturer’s instructions. Briefly, cells were trypsinized
with 0.25% trypsin, washed twice with PBS and collected by
centrifugation (192 × g, 5 min). Cells were resuspended with
binding buffer at a density of 1×106/ml, stained with
Annexin V-FITC and PI for 15 min in the dark at room temperature
and analyzed by flow cytofluorometry (Guava Easycyte 5HT).
Cell viability analysis
MTT assays were performed to assess the sensitivity
of cells to drugs. Briefly, glioma cells were seeded at a density
of 3,000 cells/well in 96-well microplates. The following day,
cells were treated with TMZ and/or compound C for 72 h. Following
culture, 20 μl MTT (5 mg/ml) was added to each well, and plates
were placed at 37°C for 4 h. DMSO (100 μl) was added to each well
to lyse the cells. Absorbance was measured at 570 nm using a
microplate spectrophotometer (Thermo Scientific Microplate Reader;
Thermo Fisher Scientific Inc., Waltham, MA, USA).
Statistical analysis
All experiments were performed in triplicate, and
results are presented as the mean ± standard deviation. Statistical
analysis of the data was performed using Student’s t-test (for two
groups) or one-way analysis of variance (for three or more groups).
P<0.05 and P<0.01 were considered to indicate a statistically
significant difference.
Results
TMZ treatment induces autophagy in
glioma
Induction of autophagy by TMZ has been documented in
glioma cell lines and surgical specimens (11,12).
In agreement with this, the current results showed that treatment
with 100 μM TMZ induced autophagy as shown by a significant
increase of AVOs and enhanced cleavage of LC3B (Fig. 1A and B). ATM, AMPK and ULK1 were
activated, as shown by western blot analysis, following TMZ
treatment (Fig. 1B).
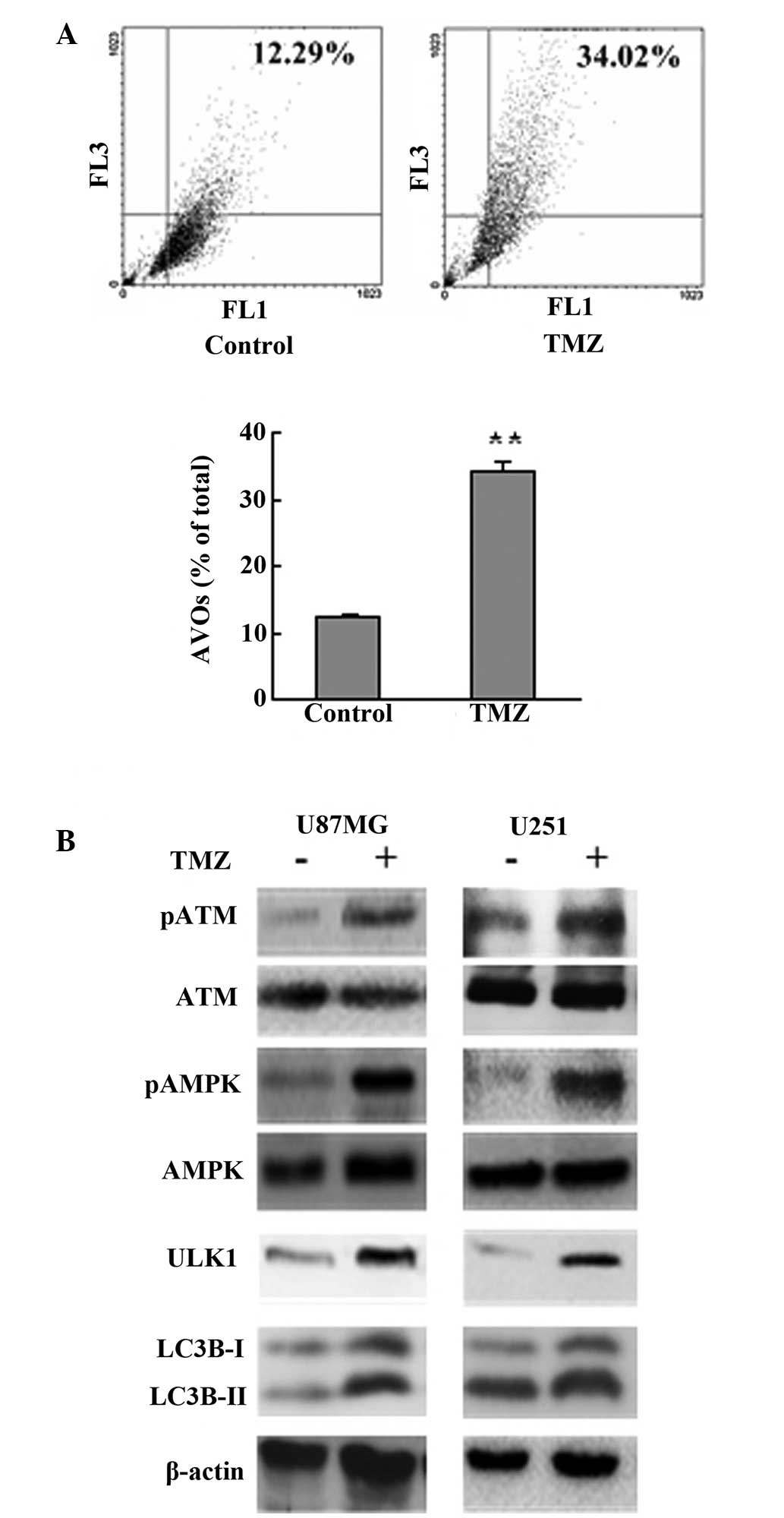 | Figure 1TMZ treatment induced autophagy in
glioma. (A) U87MG cells were treated with vehicle or TMZ (100 μM)
as indicated for 72 h. Following drug treatment, acridine orange
was added at a final concentration of 1 μg/ml for a period of 15
min. Next, cells were collected and AVOs were detected by flow
cytometry. A total of 5,000 ungated events were analyzed. (B) U87MG
and U251 glioma cells were treated with TMZ (100 μM) for 72 h.
Next, cells were harvested, and analyzed by western blot analysis.
Vehicle was used as a negative control. β-actin was used as a
loading control. Data are shown as the mean ± standard deviation.
*P<0.05 and **P<0.01, vs. control
groups, n=3 for each group. TMZ, temozolomide; AVOs, acidic
vesicular organelles ATM, ataxia-telangiectasia mutated; AMPK,
adenosine monophosphate-activated protein kinase. |
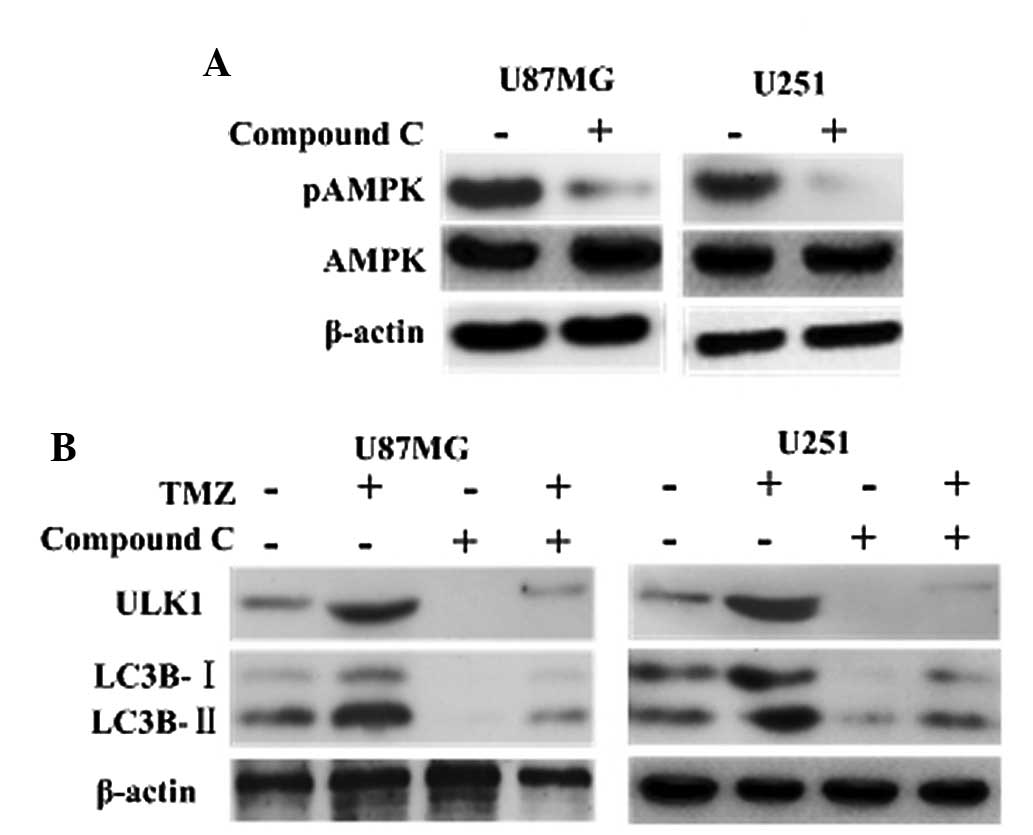
TMZ induces autophagy via AMPK-ULK1
pathways
It has been documented that AMPK is involved in the
initiation of autophagosome formation by interacting with mammalian
autophagy-initiating kinase ULK1 (18,19).
Consistently, the current results showed that inhibition of AMPK
with compound C led to depression of ULK1, and that the LC3B
cleavage was decreased significantly in the TMZ + compound C groups
compared with the other groups (Fig.
2). This indicated that TMZ induced autophagy via AMPK-ULK1
pathways.
TMZ-induced autophagy and AMPK-ULK1
activation are ATM dependent
To determine the role of ATM in the autophagy
process and AMPK-ULK1 activation, a specific ATM inhibitor,
KU-55933U, was used to inhibit ATM phosphorylation and to assess
the level of autophagy. Western blot analysis showed that TMZ
failed to induce the phosphorylation of AMPK and ULK1 following
treatment with 10 μM KU-55933, and the expression of LC3B-I and
LC3B-II declined significantly compared with the TMZ and control
groups (Fig. 3A and B). Flow
cytometric analysis showed that inhibition of ATM phosphorylation
resulted in fewer AVOs in the TMZ + KU-55933 group compared with
the TMZ group (9.24±0.38 vs. 29.90±2.14%, respectively, P<0.001;
Fig. 3C). The levels of LC3B
cleavage and AVOs in the KU-55933 group were also lower compared
with the control group (Fig. 3B and
C). Therefore, TMZ-induced autophagy and AMPK-ULK1 activation
were ATM dependent.
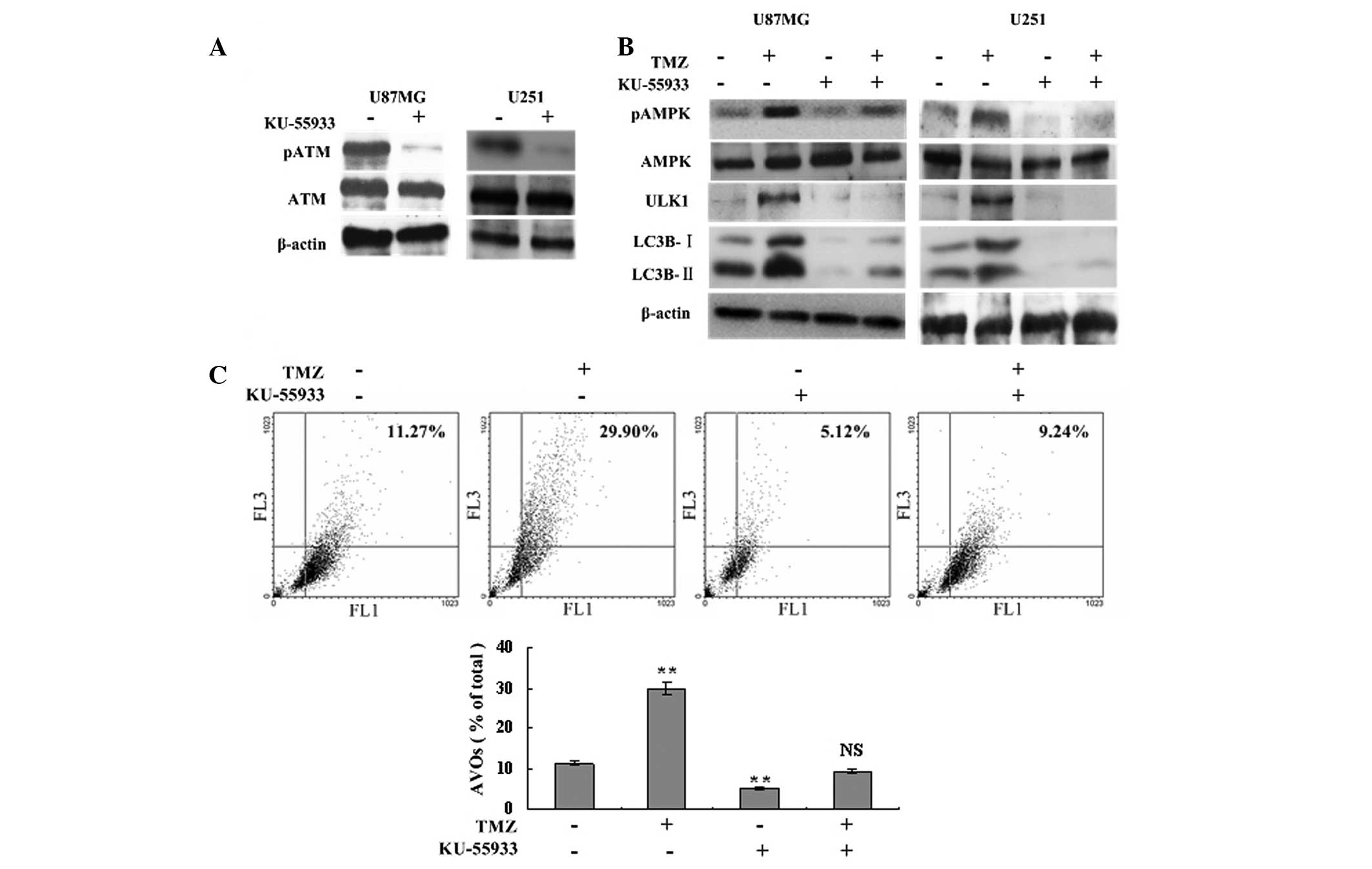 | Figure 3TMZ-induced autophagy and AMPK-ULK1
activation are ATM dependent. (A) U87MG and U251 glioma cells were
treated with 10 μM KU-55933 or vehicle. Following 72 h, cells were
harvested and analyzed by western blot analysis to determine the
effect of KU-55933 on ATM (Ser1981) phosphorylation. β-actin was
used as a loading control. (B) U87MG and U251 cells were divided
into four groups, and treated with vehicle, TMZ (100 μM), KU-55933
(10 μM) and TMZ (100 μM) + KU-55933 (10 μM) for 72 h, respectively.
Subsequently, cells were collected and subjected to western blot
analysis with antibodies against phosphorylated AMPKα (Thr172),
AMPKα, phosphorylated ULK1 (Ser467) and LC3B. β-actin was used as a
loading control. (C) U87MG cells were treated with vehicle, TMZ
(100 μM) and/or KU-55933 (10 μM) as indicated for 72 h. Following
drug treatment, acridine orange was added at a final concentration
of 1 μg/ml for a period of 15 min. Next, cells were collected and
AVOs were detected with flow cytometry. A total of 5,000 ungated
events were analyzed. Data are shown as the mean ± standard
deviation. *P<0.05 and **P<0.01, vs.
the control groups, n=3 for each group. NS not statistically
significant; TMZ, temozolomide; AVOs, acidic vesicular organelles;
AMPK, adenosine monophosphate-activated protein kinase; ATM,
ataxia-telangiectasia mutated. |
Inhibition of AMPK augmented the
cytotoxicity of TMZ by disrupting autophagy
Since autophagy serves as a cytoprotective process
and ATM mutation results in ataxia telangiectasia, the AMPK
inhibitor compound C was employed to investigate the effect of
autophagy disruption on TMZ cytotoxicity from a possible medical
treatment standpoint. AMPK inhibition with compound C was observed
to interrupt TMZ-induced autophagy (Fig. 2). In the current study, compound C
was found to augment TMZ-induced DNA damage, as indicated by
increased γH2AX detected in the TMZ + compound C group compared
with the control, TMZ and compound C groups in U87MG and U251 cells
(Fig. 4A). MTT analysis also
showed that the TMZ + compound C group exhibited slower growth
compared with that of the other groups (Fig. 4B). Next, the level of apoptosis in
each group was assessed with Annexin V-FITC/PI double staining. The
levels of apoptotic cells in the control, TMZ, compound C and TMZ +
compound C groups were observed to be 6.52, 13.32, 10.69 and
25.02%, respectively (Fig. 4C).
These results indicated that inhibition of AMPK augmented the
cytotoxicity of TMZ by interrupting autophagy.
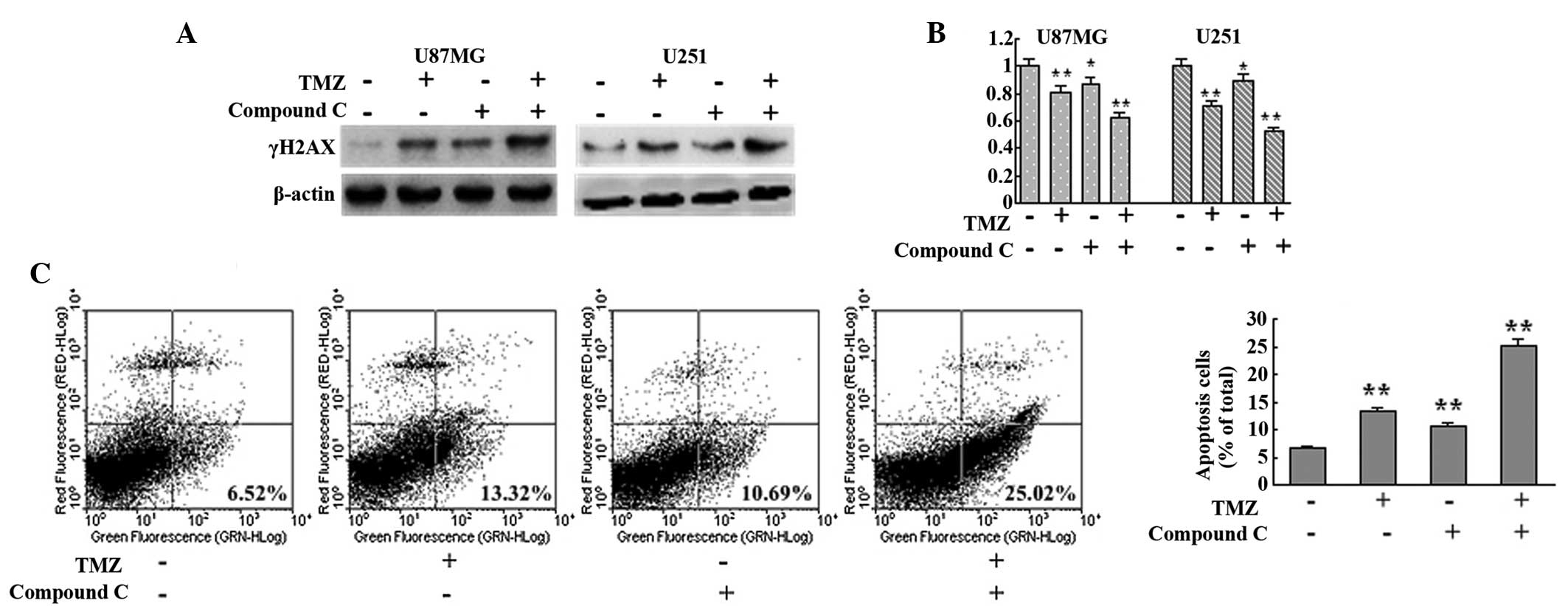 | Figure 4Inhibition of AMPK with compound C
augments the cytotoxicity of TMZ. (A) U87MG and U251 cells were
treated with vehicle, TMZ (100 μM), compound C (5 μM) or TMZ (100
μM) + compound C (5 μM) for 72 h. Next, cells were collected and
subjected to western blot analysis to detect the expression of
γH2AX. β-actin was used as a loading control. (B) U87MG and U251
cells were seeded at a density of 3,000 cells/well in 96-well
microplates and treated with vehicle, TMZ (100 μM), compound C (5
μM) or TMZ (100 μM) + compound C (5 μM) for 72 h. Following
culture, MTT assays were performed to assess the cell viability
(n=6 for each group). (C) U87MG cells were treated with vehicle,
TMZ (100 μM), compound C (5 μM) or TMZ (100 μM) + compound C (5 μM)
for 72 h. Subsequently, cells were collected and apoptosis was
detected with Annexin-V-fluorescein isothiocyanate/propidium iodide
double staining (n=3 for each group). Data are shown as the mean ±
standard deviation. *P<0.05 and
**P<0.01, vs. control group. AMPK, adenosine
monophosphate-activated protein kinase; TMZ, temozolomide. |
Discussion
Autophagy is a highly conserved catabolic process in
which cells self-digest organelles and other macromolecules via the
autophagosome. Autophagy ameliorates the negative effects of
dysregulated cellular metabolism, allowing a steady supply of
nutrients and removal of damaged organelles (20–22).
TMZ induces an autophagy-associated adenosine triphosphate (ATP)
surge through the degradation of cellular proteins and organelles,
which maintains cellular homeostasis and survival. In addition,
inhibition of autophagy augments TMZ-induced apoptosis (12,23).
The current results also showed that 100 μM TMZ treatment induced
autophagy in U87MG and U251 glioma cells. However, the molecular
mechanisms by which TMZ induces autophagy remain largely
unknown.
AMPK is a conserved sensor of intracellular energy,
which is activated in response to low nutrient availability and
cellular stress, and is involved in the initiation of autophagosome
formation by interacting with mammalian autophagy-initiating kinase
ULK1 (18,19). The current results showed that TMZ
treatment led to significant AMPK phosphorylation and ULK1
activation during the process of autopahgy. When AMPK
phosphorylation was inhibited by compound C, TMZ-induced autophagy
was significantly interrupted as indicated by a decrease in LC3B
cleavage. Next, the mechanism of AMPK-ULK1 pathway activation was
investigated.
ATM kinase forms a central node in the DNA damage
response phosphorylation cascade by contributing to the initiation,
amplification and transmission of the DNA damage signal to
downstream substrates (7,24). The results indicated that AMPK-ULK1
pathways were one of these downstream pathways. TMZ failed to
induce AMPK-ULK1 activation following KU-55933 treatment, which led
to a decrease of LC3B cleavage and AVO formation. Thus, TMZ-induced
DNA foci recruit and activate ATM kinase, which in turn evokes
phosphorylation of AMPK-ULK1 and subsequently elicits autophagy.
Thus, glioma cells may supply steady nutrients and energy for DNA
damage repair or other cellular processes.
Based on this hypothesis, interruption of
ATM-AMPK-ULK1 pathways results in autophagy inhibition and augment
TMZ cytotoxicity. To examine this hypothesis, autophagy was
interrupted with the AMPK inhibitor compound C, and AMPK inhibition
was found to augment TMZ cytotoxicity as observed by impaired cell
viability, an increase of γH2AX-marked DSBs and elevated numbers of
apoptotic U87MG cells. We hypothesize that AMPK inhibition
interrupts the cytoprotective process of autophagy, which results
in augmentation of the TMZ cytotoxic effect and promotes glioma
cell death under apoptotic stress. These results suggest that
glioma chemoresistance may be overwhelmed by targeting AMPK,
particularly for MGMT-negative patients.
In conclusion, TMZ treatment induces autophagy
through ATM-AMPK-ULK1 pathways, and AMPK inhibition augments TMZ
cytotoxicity. The current results suggest that AMPK may be a
treatment target to overwhelm TMZ chemoresistance.
Acknowledgements
The authors would like to acknowledge financial
supports from the China Postdoctoral Science Foundation (grant no.
2012M512182), the Guangdong Natural Science Foundation (grant no.
S2012040006588) and the Guangzhou Science and Technology Project
(grant no. 201300000150).
References
|
1
|
Schwartzbaum JA, Fisher JL, Aldape KD and
Wrensch M: Epidemiology and molecular pathology of glioma. Nat Clin
Pract Neurol. 2:494–503. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stupp R, Mason WP, van den Bent MJ, et al:
Radiotherapy plus concomitant and adjuvant temozolomide for
glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Caporali S, Falcinelli S, Starace G, et
al: DNA damage induced by temozolomide signals to both ATM and ATR:
role of the mismatch repair system. Mol Pharmacol. 66:478–491.
2004.PubMed/NCBI
|
|
4
|
Zhang J, Stevens MF and Bradshaw TD:
Temozolomide: mechanisms of action, repair and resistance. Curr Mol
Pharmacol. 5:102–114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu J, Zhang X, Zhang L, et al: Skp2 E3
ligase integrates ATM activation and homologous recombination
repair by ubiquitinating NBS1. Mol Cell. 46:351–361. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Andegeko Y, Moyal L, Mittelman L, Tsarfaty
I, Shiloh Y and Rotman G: Nuclear retention of ATM at sites of DNA
double strand breaks. J Biol Chem. 276:38224–38230. 2001.PubMed/NCBI
|
|
7
|
Shiloh Y and Ziv Y: The ATM protein
kinase: regulating the cellular response to genotoxic stress, and
more. Nat Rev Mol Cell Biol. 14:197–210. 2013. View Article : Google Scholar
|
|
8
|
Jiang G, Li LT, Xin Y, Zhang L, Liu YQ and
Zheng JN: Strategies to improve the killing of tumors using
temozolomide: targeting the DNA repair protein MGMT. Curr Med Chem.
19:3886–3892. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Skiriute D, Vaitkiene P, Saferis V, et al:
MGMT, GATA6, CD81, DR4, and CASP8 gene promoter methylation in
glioblastoma. BMC Cancer. 12:2182012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Felsberg J, Thon N, Eigenbrod S, et al:
Promoter methylation and expression of MGMT and the DNA mismatch
repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and
recurrent glioblastomas. Int J Cancer. 129:659–670. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Natsumeda M, Aoki H, Miyahara H, et al:
Induction of autophagy in temozolomide treated malignant gliomas.
Neuropathology. 31:486–493. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin CJ, Lee CC, Shih YL, et al: Inhibition
of mitochondria- and endoplasmic reticulum stress-mediated
autophagy augments temozolomide-induced apoptosis in glioma cells.
PLoS One. 7:e387062012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kabeya Y, Mizushima N, Yamamoto A,
Oshitani-Okamoto S, Ohsumi Y and Yoshimori T: LC3, GABARAP and
GATE16 localize to autophagosomal membrane depending on form-II
formation. J Cell Sci. 117:2805–2812. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu J, Dang Y, Su W, et al: Molecular
cloning and characterization of rat LC3A and LC3B - two novel
markers of autophagosome. Biochem Biophys Res Commun. 339:437–442.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Murrow L and Debnath J: Autophagy as a
stress-response and quality-control mechanism: implications for
cell injury and human disease. Annu Rev Pathol. 8:105–137. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Paglin S, Hollister T, Delohery T, et al:
A novel response of cancer cells to radiation involves autophagy
and formation of acidic vesicles. Cancer Res. 61:439–444.
2001.PubMed/NCBI
|
|
17
|
Graf MR, Jia W and Loria RM: The
neuro-steroid, 3beta androstene 17alpha diol exhibits potent
cytotoxic effects on human malignant glioma and lymphoma cells
through different programmed cell death pathways. Br J Cancer.
97:619–627. 2007. View Article : Google Scholar
|
|
18
|
Wong PM, Puente C, Ganley IG and Jiang X:
The ULK1 complex: sensing nutrient signals for autophagy
activation. Autophagy. 9:124–137. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sanchez AM, Csibi A, Raibon A, et al: AMPK
promotes skeletal muscle autophagy through activation of forkhead
FoxO3a and interaction with Ulk1. J Cell Biochem. 113:695–710.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Leone RD and Amaravadi RK: Autophagy: a
targetable linchpin of cancer cell metabolism. Trends Endocrinol
Metab. 24:209–217. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cheong H, Lu C, Lindsten T and Thompson
CB: Therapeutic targets in cancer cell metabolism and autophagy.
Nat Biotechnol. 30:671–678. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:651–662. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Katayama M, Kawaguchi T, Berger MS and
Pieper RO: DNA damaging agent-induced autophagy produces a
cytoprotective adenosine triphosphate surge in malignant glioma
cells. Cell Death Differ. 14:548–558. 2007. View Article : Google Scholar
|
|
24
|
Marinoglou K: The role of the DNA damage
response kinase ataxia telangiectasia mutated in neuroprotection.
Yale J Biol Med. 85:469–480. 2012.PubMed/NCBI
|