Introduction
Rheumatoid arthritis (RA) is a chronic destructive
disease (1) affecting ~1% of the
population worldwide, most commonly middle-aged females (2). RA is characterized by chronic
inflammation of the synovium, particularly of the small joints,
which commonly leads to destruction of articular cartilage and
juxta-articular bone. It is often accompanied with systemic
manifestations, including anemia, fatigue and osteoporosis. RA has
a severe impact on quality of life and so the improvement in early
diagnosis and effective therapies is urgently required.
With the development of genomics, the understanding
of susceptibility and severity of RA has rapidly advanced, and
numerous key RA-associated genes and metabolic pathways, which may
have important roles in the pathogenesis and progression of RA,
have been described. For example, the toll-like receptor (TLR)
signaling pathway has been identified to contribute to the
pathogenesis of RA (3). TLRs are a
family of type I integral membrane glycoprotein pattern recognition
receptors, which are well known for their role in the recognition
of microbial ligands and in the development of adaptive immunity
(4). TLRs are also important in
the persistence of RA and the destruction of the joint by the
chronic expression of pro-inflammatory cytokines and chemokines,
including tumor necrosis factor-α, interleukin (IL)-1β, IL-6 and
IL-8. In RA, TLRs are important for the generation of adaptive
immunity, including the activation of T and B cells. However,
instead of the normal dampening of the immune response (5) and the downregulation of innate
immunity in patients with RA, there is the persistent pathogenic
expression of inflammatory cytokines, including TNF-α, IL-1β and
IL-6, each of which have been targeted successfully in patients
with RA. Increasing evidence has demonstrated the role of TLRs in
the persistent, progressive activation of macrophages that appears
to drive this destructive inflammatory process (6). In addition, the p53 signaling
pathway, another RA-associated metabolic pathway, has also been
attracting significant attention. It is reported that
overexpression and functional mutations of the tumor suppressor p53
protein have been demonstrated in RA synovial tissue, most
extensively in patients with advanced destructive type disease
(7). Recent studies suggest that
p53 induction is a general phenomenon in inflammation, directed at
modulating normal inflammatory responses (8). A higher p53 expression has been
demonstrated in synovial tissue from patients with destructive RA
disease and the association between p53 expression and joint damage
have also been examined (9).
Numerous studies of p53 mutations and protein expression in RA and
other inflammatory disorders have consistently demonstrated an
association between p53 expression and joint damage (10–12).
Other signaling pathways involved in RA, including the NOD-like
receptor signaling pathway (10),
the T cell receptor signaling pathway (11) and the Wnt signaling pathway
(12) have also been investigated.
A number of other factors, such as inflammatory cytokines (for
example IL-6 and IL-1) (13), the
human leucocyte antigen (14) and
DNA methylation (15) have been
identified to affect the development of RA and may be used as
diagnostic markers for RA. Although the understanding of the
mechanisms of RA has improved, further studies are required to
further elucidate the pathogenesis of RA comprehensively, to
facilitate the development of novel diagnostic markers and
effective treatments.
At present, numerous conventional methods exist for
the analysis of gene expression data, including contrasting of
differentially expressed genes and clustering. However, an
increasing number of studies suggest that to reflect the
interaction between genes, only searching for genes with
significantly differential expression levels at different
conditions lacks sufficient accuracy. Rather, the identification of
a set of genes with the same expression profiles that functionally
interact in different states, may better reflect the interaction
between genes under different conditions, and this set of genes are
defined as differentially co-expressed genes (16,17).
The aim of the present study was to analyze the differentially
co-expressed genes (DCGs) and differentially co-expressed links
(DCLs) of RA and osteoarthritis based on microarray expression
data. The results may improve the understanding of the underlying
molecular mechanisms of RA and facilitate the development of novel
approaches for improving the diagnosis or treatment of RA.
Materials and Methods
Gene expression data of RA
Firstly, GSE27390 (18) chip expression data were selected
from the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/), and samples in the
chip GPL570 (HG-U133_Plus_2) Affymetrix Human Genome U133 Plus 2.0
Array platform (Affymetrix, Inc., Santa Clara, CA, USA) were
selected for analysis. There were a total of 19 samples under this
platform, among which 10 samples were from patients with RA, while
the other nine samples were from patients with osteoarthritis.
Following this, the sample data and platform annotation files were
downloaded.
Analysis of differential
co-expression
The chip data obtained were standardized by the
robust multi-array average method (19) using the Affy package of R software
(www.r-project.org). Then, the samples were divided
into two groups (RA vs. osteoarthritis) and the t-test method in
the LIMMA package (a set of tools for background correction and
scaling) of R software (20) was
used to calculate the differentially expressed genes (DEGs), with
threshold of |logFC|>1.0 and a P-value <0.05. Finally, the
DCGs and DCLs were calculated with the cutoff criterion of
q<0.25 using the functions of DCe, DCp and DCsum in the
differential coexpression analysis and differential regulation
analysis of gene expression microarray data (DCGL) package
(21,22). The DCGL package included four
modules: Gene screening, relations screening, analysis of
differential co-expression and differential regulation
analysis.
Functional enrichment analysis of
DCGs
As the Kyoto Encyclopedia of Genes and Genomes
(KEGG) pathway database (23) is a
relatively common and comprehensive database that contains a
variety of biochemical pathways, it was also selected to continue
the enrichment analysis of the DCGs screened during the analysis of
RA microarray data. This database was utilized to identify which
biochemical pathways, with their associated biological functions,
the co-expressed genes are more likely to affect during the
pathological development of RA. The online tool, the Database for
Annotation, Visualization and Integrated Discovery (DAVID)
(24) was used to conduct pathway
enrichment analysis for DCGs.
Transcriptional regulation
correlation
TRANSFAC (25) is a
database associated with transcription factors and their genomic
binding sites on DNA-binding profiles. It is composed of SITE,
GENE, FACTOR, CLASS, MATRIX, CELLS, METHOD and REFERENCE, etc. All
associations between human transcription factors and target genes
in TRANSFAC were downloaded and compiled, which contained a total
of 298 transcription factors (TF) and 6458 associations.
Construction of RA-related transcription
regulation network
The obtained differential co-expression correlations
were mapped to the associations between human TFs and their target
genes, and the TFs were corresponded to the already known target
genes. Then the transcription regulation associations of DCGs were
obtained. Finally, cytoscape was used for network description
(26).
Results
Differential co-expression analysis
Affy and LIMMA packages in R software were used to
calculate the chip data and 1,858 DEGs were obtained with the
cut-off criteria of |logFC|>1.0 and a P-value <0.05. The DCGL
method in R software was used to perform differential co-expression
analysis of the expression data of DEGs in RA to obtain a P-value
and q value corresponding to each gene, and the DCLs between genes.
Following selection of q<0.25 as the threshold for screening
DCGs and DCLs, 457 DCGs and 101,988 DCLs with significant
correlations were obtained.
Biological pathways closely associated
with RA
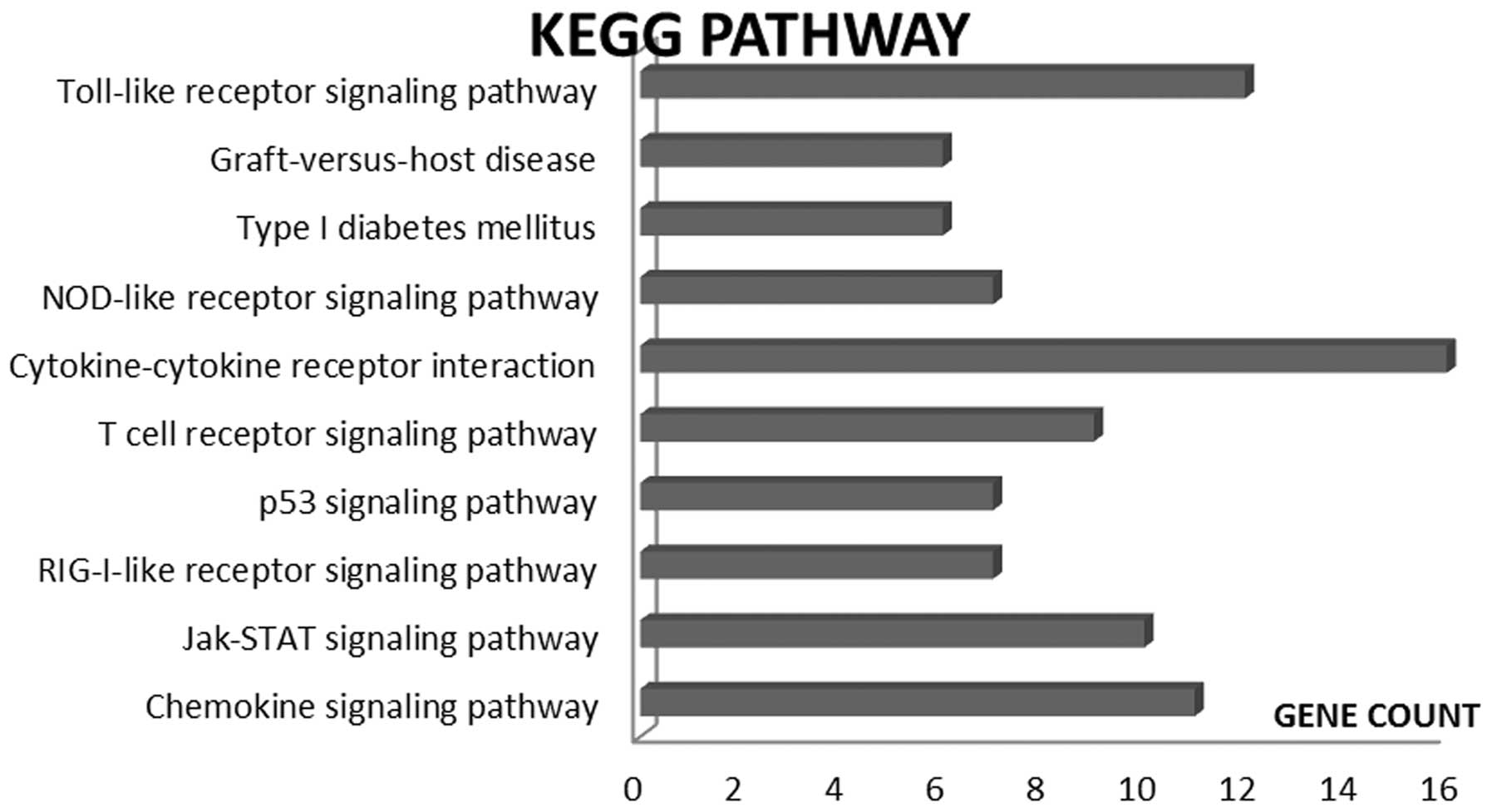
Following identification of the DCGs, the online
tool, DAVID, was used to perform KEGG pathway significant
enrichment analysis of 457 DCGs associated with RA and the ten
metabolic pathways with the smallest P-values were selected, as
revealed in Fig. 1. From this, it
was identified that the major metabolic pathways of the DCGs were
associated with the following mechanisms: TLR signaling (27,28),
graft-versus-host disease (29),
the NOD-like receptor signaling (30), cytokine-cytokine receptor
interaction, T cell receptor signaling, p53 signaling, RIG-I-like
receptor signaling, Jak-STAT signaling and chemokine signaling.
Analysis of transcriptional
regulation
The 101,988 pairs of DCLs were compared with the
known links between TFs and target genes. Six links between TFs and
target genes that are known to be involved in RA were obtained,
including STAT3-TNF, PBX1-PLAU, SOCS3-STAT3, GATA1-ETS2, ETS1-ICAM4
and CEBPE-GATA1, as summarized in Table I. These six associations and 457
DCGs contained 20 TFs, including CEBPE, EGR1, EGR2, EGR3, ERG,
ETS1, ETS2, FOS, HOXA5, IKZF1, IL6, JUN, JUNB, KLF9, PLAU, PRDM1,
RUNX1, STAT1, PBX1 and STAT3. Using text mining methods, the
literature on these 20 TFs associated with RA were investigated and
the results are summarized in Table
II. Then, the 20 TFs were mapped to the known TF-target
regulatory associations and 233 regulatory associations were
achieved. Cytoscape software was utilized to describe the
associations observed, including those between the 20 transcription
factors and 572 target genes (Fig.
2). In addition, a number of TF-target associations in the
constructed network were not within DCLs when TFs and target genes
were DCGs, which indicated that the TFs and target genes may have
acted synergistically with other factors (Table III).
 | Table IThe known transcriptional regulation
associations contained in DCLs. |
Table I
The known transcriptional regulation
associations contained in DCLs.
| TF | Target | cor. 1 | cor. 2 | Type | cor. diff |
|---|
| STAT3 | TNF | 0.014514 | 0.723764 | same signed | 0.709251 |
| PBX1 | PLAU | −0.74973 | −0.13143 | same signed | 0.618294 |
| SOCS3 | STAT3 | 0.033164 | 0.628008 | same signed | 0.594844 |
| GATA1 | ETS2 | −0.114 | −0.66228 | same signed | 0.548279 |
| ETS1 | ICAM4 | −0.53926 | −0.04391 | same signed | 0.495352 |
| CEBPE | GATA1 | −0.57329 | −0.09175 | same signed | 0.481536 |
| Table IIText mining results of TFs. |
Table II
Text mining results of TFs.
|
TF |
|---|
|
|
|---|
| IL-6 | STAT3 | FOS | STAT1 | RUNX1 | EGR1 | ERG | ETS1 | JUNB | PRDM1 | EGR2 | ETS2 |
|---|
| Count | 211 | 132 | 131 | 73 | 19 | 18 | 16 | 8 | 7 | 7 | 3 | 2 |
| Table IIITFs and target genes in DCGs. |
Table III
TFs and target genes in DCGs.
| TF | Target gene |
|---|
| EGR1 | FGFR3, FGFR3, IFNG,
TNG, PLAU |
| ETS1 | OAS2, SPRY2, CCL3,
SOCS1, MDM2, IFNG, TNG, PLAU, OAS2 |
| ETS2 | GBA, CD163, MDM2,
ERG, JUNB, PLAU |
| FOS | PLAU, CCL4,
IFNG |
| IL6 | PLAU |
| JUN | TNF, PLAU |
| JUNB | PLAU |
| STAT1 | PLAU, GBP1, PSMB9,
ISG15, IFI6, IRF7, TAP1, SOCS3, IFNG |
Discussion
RA is a disease characterized by chronic
inflammatory processes that targets the synovial lining of
diarthrodial joints. Investigating the mechanisms underlying RA
development has been the focus of numerous studies, in an attempt
to improve the disabling impact RA has on quality of life. In the
present study, a DCGL method was utilized to perform a differential
co-expression analysis of microarray data of RA and a total of 457
DCGs and 101,988 DCLs were obtained. From significant enrichment
analysis of the KEGG pathway for DCGs, it was identified that the
major metabolic pathways associated with these DCGs were as
follows: The TLR signaling pathway, graft-versus-host disease, the
NOD-like receptor signaling pathway, the cytokine-cytokine receptor
interaction, the T cell receptor signaling pathway, the p53
signaling pathway, the RIG-I-like receptor signaling pathway, the
Jak-STAT signaling pathway and the chemokine signaling pathway. The
six TF-target associations included STAT3-TNF, PBX1-PLAU,
SOCS3-STAT3, GATA1-ETS2, ETS1-ICAM4 and CEBPE-GATA1. Among which,
the focus of the present study was SOCS.
SOCS family proteins consists of SOCS1-SOCS7 and
CIS1, with SOCS1, SOCS2, SOCS3 and CIS1 being the
best-characterized members of the family. Once expressed, they
inhibit the JAK-STAT pathway by several mechanisms, including the
suppression of JAK catalytic activity and prevention of STAT
recruitment to activated cytokine receptors. Evidence has revealed
that SOCS proteins are important regulators of immune and
inflammatory responses in vivo (31,32).
SOCS proteins also appear to regulate the development and
progression of arthritis. Egan et al (33) demonstrated increased arthritis
severity in mice lacking SOCS1 and interferon-γ, and STAT1
gene-knockout mice demonstrated exacerbated zymosan-induced
arthritis, possibly due to a reduction in SOCS1 (34). Furthermore, overexpression of SOCS
has inhibitory effects on arthritis, as demonstrated by the ability
of adenoviral-mediated induction of SOCS3 to markedly reduce the
severity of collagen-induced arthritis (35). Isomäki et al (35) demonstrated increased SOCS3 mRNA
expression in synovial tissues from patients with RA when compared
with those with osteoarthritis, which indicates SOCS3 is generally
upregulated in RA. Furthermore, in patients with long-standing RA,
increased levels of SOCS1 and SOCS3 may suppress cytokine signaling
and potentially prevent the deleterious actions of pro-inflammatory
cytokines. In general, upregulation of SOCS expression may
significantly affect cellular responsiveness to cytokines in
chronic RA and may be involved in disease progression by inducing
unresponsiveness to anti-inflammatory cytokines.
It was observed that the target gene of SOC3 was
STAT3, however STAT3 was also a TF in another TF-target
association, and its target gene was TNF. STAT3 has been
demonstrated to have an important role in cellular proliferation
and anti-apoptotic mechanisms (36). Evidence reveals that
hyperactivation of STAT3 may be involved in proliferation and/or
prevent apoptosis of RA synoviocytes. In addition, STAT3 activation
is identified exclusively in synovial tissue from RA, but not
osteoarthritis patients. Therefore, STAT3 may have an important
role in the mechanisms associated with RA. Furthermore, although
the cause of RA remains unclear, it has been suggested that
cytokines, particularly pro-inflammatory cytokines, including
TNF-α, IL-1 and IL-6 derived from activated synovial cells, are
important in the pathology of the disease (37). Among these cytokines, TNF-α has
been the most extensively investigated as a target in the treatment
of RA. TNF is the target gene of STAT3, which further confirms the
role of STAT3-TNF in RA. Several studies have demonstrated that
anti-TNF-α mAbs markedly ameliorate joint involvement in the
majority of patients with RA (38,39).
It is also of note that IL-6, a major target gene of TNF-α, has
been proposed to contribute to the development of arthritis, and
may induce proliferation of synovial fibroblastic cells (40) and the formation of osteoclasts in
the presence of soluble IL-6 receptors (41).
In conclusion, the six TF-target associations in
DCLs and the obtained TFs may have important roles in the
pathogenesis of RA. However, the number of samples in the present
study were limited. Therefore, whether the obtained TFs may be used
as biomarkers and whether they are a cause or consequence of the
disease remains to be elucidated in a larger prospective study.
References
|
1
|
Raza K: The Michael Mason prize: early
rheumatoid arthritis - the window narrows. Rheumatology (Oxford).
49:406–410. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nell V, Machold K, Eberl G, Stamm T,
Uffmann M and Smolen J: Benefit of very early referral and very
early therapy with disease-modifying anti-rheumatic drugs in
patients with early rheumatoid arthritis. Rheumatology (Oxford).
43:906–914. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bigelow RL, Jen EY, Delehedde M, Chari NS
and McDonnell TJ: Sonic hedgehog induces epidermal growth factor
dependent matrix infiltration in HaCaT keratinocytes. J Invest
Dermatol. 124:457–465. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Iwasaki A and Medzhitov R: Regulation of
adaptive immunity by the innate immune system. Science.
327:291–295. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hoebe K, Janssen E and Beutler B: The
interface between innate and adaptive immunity. Nat Immunol.
5:971–974. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wijbrandts CA, Vergunst CE, Haringman JJ,
Gerlag DM, Smeets TJ and Tak PP: Absence of changes in the number
of synovial sublining macrophages after ineffective treatment for
rheumatoid arthritis: implications for use of synovial sublining
macrophages as a biomarker. Arthritis Rheum. 56:3869–3871. 2007.
View Article : Google Scholar
|
|
7
|
Firestein GS, Nguyen K, Aupperle KR, Yeo
M, Boyle DL and Zvaifler NJ: Apoptosis in rheumatoid arthritis: p53
overexpression in rheumatoid arthritis synovium. Am J Pathol.
149:2143–2151. 1996.PubMed/NCBI
|
|
8
|
Yamanishi Y, Boyle DL, Pinkoski MJ, et al:
Regulation of joint destruction and inflammation by p53 in
collagen-induced arthritis. Am J Pathol. 160:123–130. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Salvador G, Sanmarti R, Garcia-Peiró A,
Rodríguez-Cros J, Muñoz-Gómez J and Cañete J: p53 expression in
rheumatoid and psoriatic arthritis synovial tissue and association
with joint damage. Ann Rheum Dis. 64:183–187. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McGovern DP, Hysi P, Ahmad T, et al:
Association between a complex insertion/deletion polymorphism in
NOD1 (CARD4) and susceptibility to inflammatory bowel disease. Hum
Mol Genet. 14:1245–1250. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Porcelli S, Yockey CE, Brenner MB and Balk
SP: Analysis of T cell antigen receptor (TCR) expression by human
peripheral blood CD4-8-alpha/beta T cells demonstrates preferential
use of several V beta genes and an invariant TCR alpha chain. J Exp
Med. 178:1–16. 1993. View Article : Google Scholar
|
|
12
|
Sen M: Wnt signalling in rheumatoid
arthritis. Rheumatology (Oxford). 44:708–713. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Deon D, Ahmed S, Tai K, et al: Cross-talk
between IL-1 and IL-6 signaling pathways in rheumatoid arthritis
synovial fibroblasts. J Immunol. 167:5395–5403. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Feldmann M and Maini RN; Lasker Clinical
Medical Research Award. TNF defined as a therapeutic target for
rheumatoid arthritis and other autoimmune diseases. Nat Med.
9:1245–1250. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nakano K, Whitaker JW, Boyle DL, Wang W
and Firestein GS: DNA methylome signature in rheumatoid arthritis.
Ann Rheum Dis. 72:110–117. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee HK, Hsu AK, Sajdak J, Qin J and
Pavlidis P: Coexpression analysis of human genes across many
microarray data sets. Genome Res. 14:1085–1094. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stuart JM, Segal E, Koller D and Kim SK: A
gene-coexpression network for global discovery of conserved genetic
modules. Science. 302:249–255. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee H-M, Sugino H, Aoki C, et al: Abnormal
networks of immune response-related molecules in bone marrow cells
from patients with rheumatoid arthritis as revealed by DNA
microarray analysis. Arthritis Res Ther. 13:R892011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Irizarry RA, Hobbs B, Collin F, et al:
Exploration, normalization, and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar
|
|
20
|
Diboun I, Wernisch L, Orengo CA and
Koltzenburg M: Microarray analysis after RNA amplification can
detect pronounced differences in gene expression using limma. BMC
Genomics. 7:2522006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu H, Liu B-H, Ye ZQ, Li C, Li YX and Li
YY: Link-based quantitative methods to identify differentially
coexpressed genes and gene pairs. BMC Bioinformatics. 12:3152011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu BH, Yu H, Tu K, Li C, Li YX and Li YY:
DCGL: an R package for identifying differentially coexpressed genes
and links from gene expression microarray data. Bioinformatics.
26:2637–2638. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kanehisa M and Goto S: KEGG: kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang da W, Sherman BT, Tan Q, Collins JR,
Alvord G, Roayaei J, Stephens R, Baseler MW, Lane HC and Lempicki
RA: The DAVID Gene Functional Classification Tool: a novel
biological module-centric algorithm to functionally analyze large
gene lists. Genome Biol. 8:R1832007.
|
|
25
|
Wingender E, Chen X, Hehl R, et al:
TRANSFAC: an integrated system for gene expression regulation.
Nucleic Acids Res. 28:316–319. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Smoot ME, Ono K, Ruscheinski J, Wang P-L
and Ideker T: Cytoscape 2.8: new features for data integration and
network visualization. Bioinformatics. 27:431–432. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang ML, Huang ZZ and Zheng Y: Estimates
and prediction on incidence, mortality and prevalence of breast
cancer in China, 2008. Zhonghua Liu Xing Bing Xue Za Zhi.
33:1049–1051. 2012.(In Chinese).
|
|
28
|
Hu F, Mu R, Zhu J, et al: Hypoxia and
hypoxia-inducible factor-1α provoke toll-like receptor
signalling-induced inflammation in rheumatoid arthritis. Ann Rheum
Dis. May 3–2013.(Epub ahead of print).
|
|
29
|
Xue F, Zhang C, He Z, Ding L and Xiao H:
Analysis of critical molecules and signaling pathways in
osteoarthritis and rheumatoid arthritis. Mol Med Rep. 7:603–607.
2013.PubMed/NCBI
|
|
30
|
Plantinga TS, Fransen J, Knevel R, et al:
Role of NOD1 polymorphism in susceptibility and clinical
progression of rheumatoid arthritis. Rheumatology (Oxford).
52:806–814. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Alexander WS: Suppressors of cytokine
signalling (SOCS) in the immune system. Nat Rev Immunol. 2:410–416.
2002.PubMed/NCBI
|
|
32
|
Kubo M, Hanada T and Yoshimura A:
Suppressors of cytokine signaling and immunity. Nat Immunol.
4:1169–1176. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Egan PJ, Lawlor KE, Alexander WS and Wicks
IP: Suppressor of cytokine signaling-1 regulates acute inflammatory
arthritis and T cell activation. J Clin Invest. 111:915–924. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
de Hooge AS, van de Loo FA, Koenders MI,
et al: Local activation of STAT-1 and STAT-3 in the inflamed
synovium during zymosan-induced arthritis: exacerbation of joint
inflammation in STAT-1 gene-knockout mice. Arthritis Rheum.
50:2014–2023. 2004.PubMed/NCBI
|
|
35
|
Isomäki P, Alanärä T, Isohanni P, et al:
The expression of SOCS is altered in rheumatoid arthritis.
Rheumatology (Oxford). 46:1538–1546. 2007.PubMed/NCBI
|
|
36
|
Nakajima K, Yamanaka Y, Nakae K, et al: A
central role for Stat3 in IL-6-induced regulation of growth and
differentiation in M1 leukemia cells. EMBO J. 15:3651–3658.
1996.PubMed/NCBI
|
|
37
|
Feldmann M, Brennan FM and Maini RN: Role
of cytokines in rheumatoid arthritis. Annu Rev Immunol. 14:397–440.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Elliott MJ, Maini R, Feldmann M, et al:
Randomised double-blind comparison of chimeric monoclonal antibody
to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid
arthritis. Lancet. 344:1105–1110. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Elliott MJ, Maini R, Feldmann M, et al:
Repeated therapy with monoclonal antibody to tumour necrosis factor
alpha (cA2) in patients with rheumatoid arthritis. Lancet.
344:1125–1127. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mihara M, Moriya Y, Kishimoto T and Ohsugi
Y: Interleukin-6 (IL-6) induces the proliferation of synovial
fibroblastic cells in the presence of soluble IL-6 receptor. Br J
Rheumatol. 34:321–325. 1995. View Article : Google Scholar
|
|
41
|
Tamura T, Udagawa N, Takahashi N, et al:
Soluble interleukin-6 receptor triggers osteoclast formation by
interleukin 6. Proc Natl Acad Sci USA. 90:11924–11928. 1993.
View Article : Google Scholar : PubMed/NCBI
|