Introduction
Tumor invasion and metastasis are the main
characteristics of various types of aggressive human cancer,
including breast cancer (1). Cell
invasion is one of the initiation steps for the metastatic cascade,
during which cancer cells migrate through the extracellular matrix
from the primary tumor, which is associated with the upregulated
expression of matrix metalloproteinases (MMPs) (2–4).
Therefore, it is important to elucidate the mechanisms underlying
cancer invasion.
Histone acetylation status regulated by histone
acetyltransferases (HATs) and histone deacetylases (HDACs) is
important in the regulation of gene expression by affecting
chromatin structure and accessibility (5,6).
HDACs are recruited to DNA-bound transcription factors resulting in
the removal of acetyl groups from nucleosomal histones or directly
interact with transcription factors to modulate gene expression
(7–10). HDAC inhibition leads to the
accumulation of acetylation in histones and transcription factors,
and specifically programmed gene expression patterns (11,12).
In humans, the reduction of histone acetylation is
significantly associated with tumor progression and invasion
(13). Emerging evidence indicates
that histone deacetylase inhibitors (HDACi) induce growth
inhibition, cell cycle arrest and programmed cell death in diverse
cancer cells (14,15). HDACi treatment upregulates the
expression of suppressors of metastasis and downregulates
invasion-promoting genes, resulting in the repression of cancer
cell invasion and metastasis (16). HDAC inhibition is emerging as a
potential strategy for cancer therapy and several HDACi have been
developed for clinical trials in patients with solid malignancies
(17). Trichostatin A (TSA), a
non-competitive reversible inhibitor of HDAC activity, has been
reported to inhibit cancer invasion and metastasis in vivo
and in vitro (18–20). HDACi modulate cancer progression
through affecting the acetylation of histone and non-histone
proteins to reactivate the transcription of differential target
genes. It has been proposed that TSA upregulates RECK to suppress
MMP2 activation and cancer cell invasion (21). Therefore, it is important to
investigate the targets of HDACi in order to elucidate the
molecular mechanisms underlying HDACi elicited phenotypes.
In the present study, TSA was observed to inhibit
cell invasion in MCF-7 breast cancer cells. Furthermore, the
expression of tissue inhibitors of metalloproteinase 2/3 (TIMP2/3)
was upregulated and the expression of MMP2/9 was decreased by TSA
treatment. Notably, TIMP2/3 and MMP2/9 have been revealed as
targets of TET1 in prostate and breast cancer invasion (22). Additionally, tumor development is
associated with a decrease in TET expression and 5-methylcytosine
hydroxylation (23). TIMP2 and
TET1 consistently demonstrated downregulation during breast cancer
progression in vivo. Our hypothesis was that TET1 may be one
of the targets of HDACi in breast cancer invasion. As expected,
TET1 was upregulated by TSA stimulation and TET1 knockdown
facilitated breast cancer cell invasion. Importantly, TET1
depletion impaired TSA induced suppression of cell invasion,
suggesting that TET1 may act as one of the HDACi targets partially
mediating TSA elicited anti-cancer activity.
Materials and methods
Patient samples and cell culture
The present study was approved by the ethics
committee of Shanghai Tongren Hospital (Shanghai, China), and
written informed consent was obtained from all participants prior
to the study. A total of 61 cancer specimens from breast cancer
patients from stages I to IV were collected and the conditions of
these patients are summarized in Table
I. Samples were individually fresh-frozen in TRIzol reagent
(Invitrogen Life Technologies, Carlsbad, CA, USA). All patients
were histologically examined at Shanghai Tongren Hospital
(Shanghai, China) and written informed consent was obtained from
all study participants. Breast cancer MCF-7 cells were cultured in
RPMI-1640 medium (Invitrogen Life Technologies) supplemented with
10% fetal calf serum at 37°C and 5% CO2. TSA dissolved
in DMSO was applied in the present study with the indicated
concentrations.
 | Table ICharacteristics of breast cancer
patients. |
Table I
Characteristics of breast cancer
patients.
| Age (years) |
|---|
|
|
|---|
| Phase | 40–49 | 50+ |
|---|
| I | 5 | 7 |
| II | 12 | 14 |
| III | 7 | 9 |
| IV | 3 | 4 |
Wound-healing assay
Wound healing was performed as previously described
in 12-well plates (24). Briefly,
after MCF-7 cells grew to >90% confluence, a wound was
introduced using a 200 μl pipette tip. Subsequently, the cells were
washed once with PBS and then incubated at 37°C and 5%
CO2. Wound closure was monitored over an indicated time
period and images were captured at the four intersecting edges of
the cross. Wound width at 0 and 24 or 36 h was measured and the
difference plotted as the percentage of wound closure.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from MCF-7 cells or cancer
tissues using TRIzol reagent (Invitrogen Life Technologies). For
each sample, 2.5 or 5 μg of total RNA was reverse transcribed using
the QuantiTect Reverse Transcription kit (Qiagen, Hilden, Germany).
The cDNA product was then quantified by SYBR-Green real-time PCR
master mix (Toyobo, Osaka, Japan). The primers used were as
follows: TET1, forward 5′-GAGCCTGTTCCTCGATGTGG-3′ and reverse
5′-CAAACCCACCTGAGGCTGTT-3′; GAPDH, forward
5′-GTGTTCCTACCCCCAATGTGT-3′ and reverse
5′-ATTGTCATACCAGGAAATGAGCTT-3′; TIMP2, forward
5′-GGGTCTCGCTGGACATTG-3′ and reverse 5′-TTGATGTTCTTCTCCGTGACC-3′;
TIMP3, forward 5′-CATGTGCAGTACATCCATACGG-3′ and reverse
5′-CATCATAGACGCGACCTGTCA-3′; MMP2, forward
5′-AAGGCCAAGTGGTCCGTGTGAA-3′ and reverse
5′-AACAGTGGACATGGCGGTCTCAG-3′; MMP9, forward
5′-CACGTCCACCCCTCAGAGC-3′ and reverse
5′-GCCACTTGTCGGCGATAAGC-3′.
Western blot analysis
MCF-7 cells were lysed with two-fold loading buffer
[20 mM of Tris-HCl (pH 7.4), 2 mM of EDTA and 1% Triton X-100] and
the supernatant was subjected to western blot analysis, which was
performed as previously described (25). GAPDH was used as the loading
control. The following primary antibodies were used: anti-TET1
(Abcam, Cambridge, UK; 1:500), anti-MMP2 (Abcam; 1:400), anti-TIMP2
(Abcam; 1:500) and anti-GAPDH (Millipore, Billerica, MA, USA;
1:10,000).
shRNA and transfection
Chemically synthesized TET1 shRNA and scramble shRNA
(control) were annealed and cloned into a short interfering RNA
expressing vector named pSUPER (Oligoengine, Seattle, WA, USA).
Control or TET1 shRNA targeted sequences were: control shRNA
5′-GCTACGAAGCACCTCTCTTAG-3′ and TET1 shRNA
5′-CGATGCAAGCCATCCTTTCGA-3′. Plasmids purified by the Qiagen
purification kit were transfected into MCF-7 cells at 40–60%
confluency with Lipofectamine 2000 (Invitrogen Life Technologies)
according to the manufacturer’s instructions.
Statistic analysis
SPSS 11.0 software was used for statistical analysis
in the present study. The values are presented as the mean ± SD and
one-way ANOVA was applied for group differences. All tests were
two-sided and P<0.05 was considered to indicate a statistically
significant difference.
Results
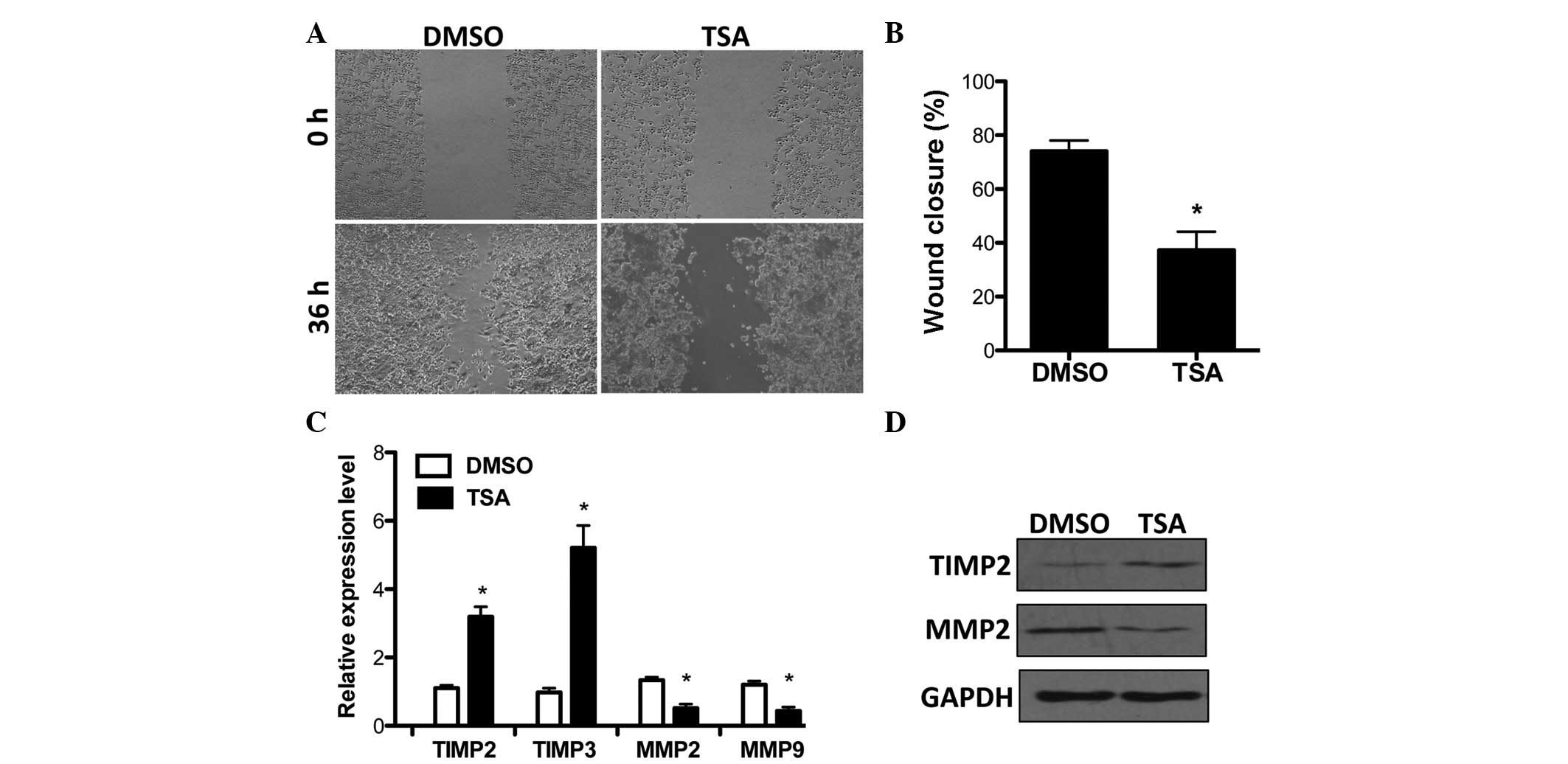
HDACi suppresses breast cancer cell
invasion and regulates TIMP2/3 and MMP2/9 expression
Previously, HDACi were reported to inhibit breast
cancer cell invasion (18,26). Prior to examining the mechanisms of
HDAC in cancer invasion, the effects of TSA on cell invasion were
examined by a wound-healing assay in MCF-7 breast cancer cells.
Consistent with previous findings, TSA significantly suppressed
cell invasion (Fig. 1A and B).
TIMP2 and 3, the important suppressors of cancer cell invasion
(27,28), were upregulated under the
stimulation of TSA. By contrast, the expression of MMP2/9, which
was correlated with clinicopathological disease variables in breast
cancer (29,30), was increased by HDAC inhibition
(Fig. 1C). Furthermore, TSA
affected MMP2 and TIMP2 expression and this was verified at the
protein level (Fig. 1D). In
addition, MMP2/9 and TIMP2/3 mRNA levels were not affected when TSA
was administered to cells within 2 h (data not shown). This
demonstrates that TIMP2/3 and MMP2/9 expression may be indirectly
modulated by HDAC in breast cancer.
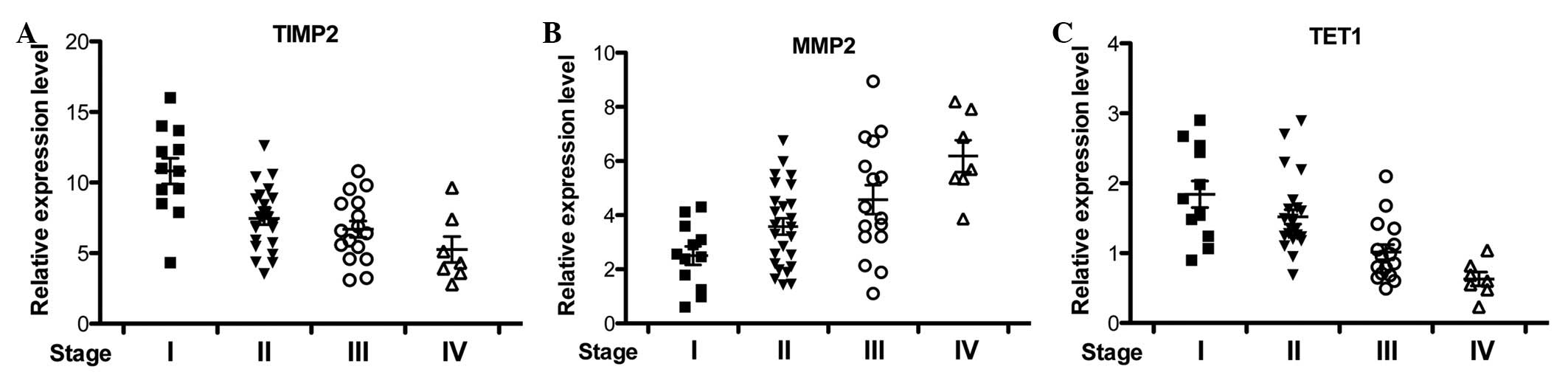
TIMP2, MMP2 and TET1 expression during
breast cancer development
Coincidently, TIMPs and MMPs were revealed as
downstream targets of TET1 and TET1 suppresses invasion partly
through TIMP activation and MMP inhibition in prostate and breast
cancer (22). In order to
investigate the correlation between TIMP2, MMP2 and TET1 in
vivo, the expression of these genes in multiple breast cancer
patients at different stages was examined by qRT-PCR. TIMP2 and
MMP2 expression was progressively downregulated and upregulated
with breast cancer development, respectively (Fig. 2A and B). Consistently, TET1 mRNA
level was reduced in breast cancer tissues (Fig. 2C), which corrrelated with TIMP2
expression changes (23).
Collectively, TIMP2 and TET1 downregulation and MMP2 upregulation
were correlated with breast cancer progression.
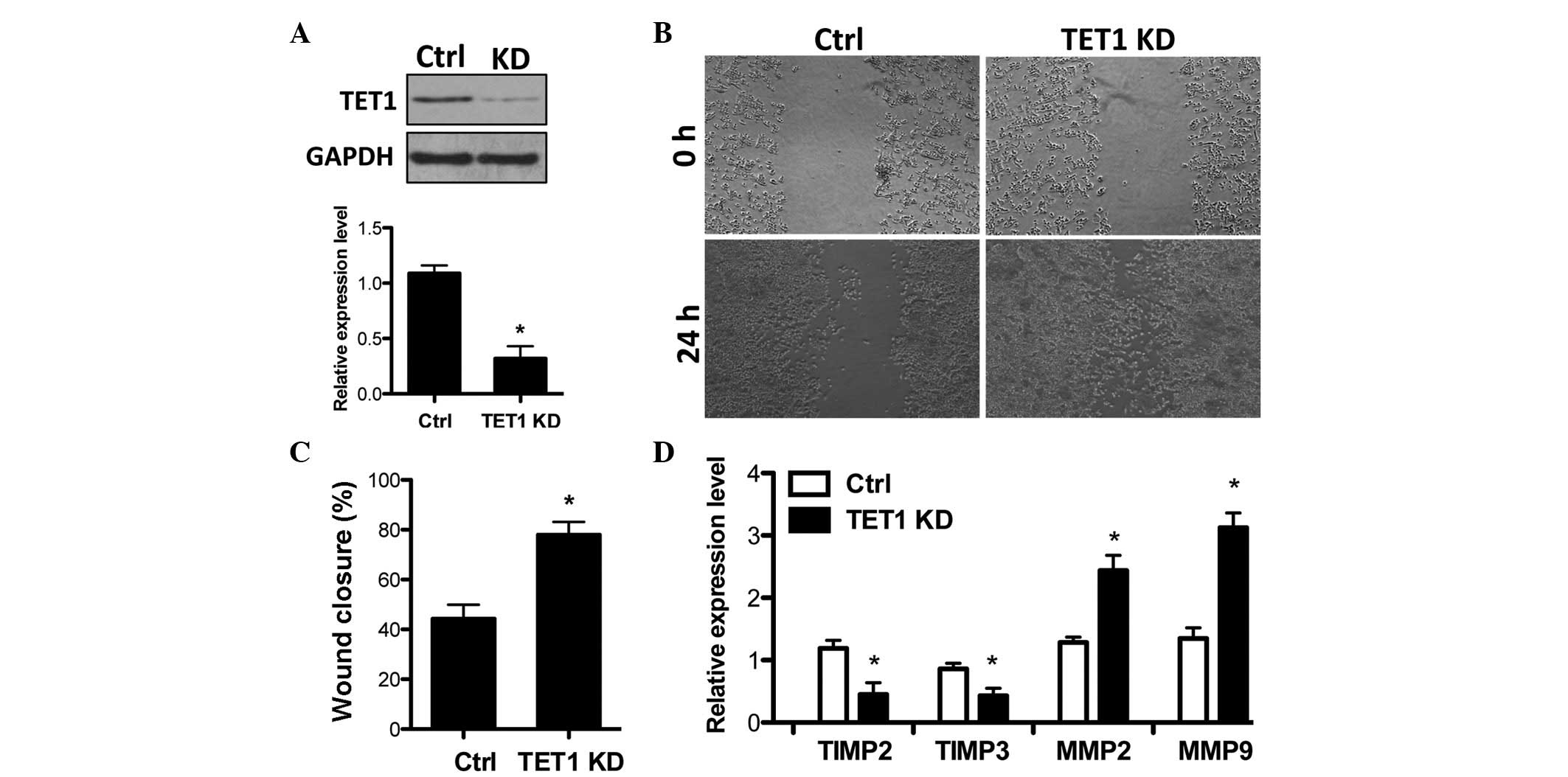
TET1 knockdown facilitates breast cancer
cell invasion
Subsequently, the present study examined whether the
reduction of TET1 is functionally involved in breast cancer cell
invasion. TET1 shRNA was delivered into MCF-7 cells to efficiently
knockdown TET1 expression (Fig.
3A). Then the effects of TET1 knockdown on cell invasion were
analyzed and the results demonstrated that TET1 depletion by TET1
shRNA increased the cell invasion capacity (Fig. 3B and C). Notably, TET1 knockdown
resulted in a decrease in TIMP2/3 and the upregulation of MMP2/9
expression (Fig. 3D), which is
opposite to the effects of TSA in MCF-7 cells (Fig. 1A). This indicates that TET1
suppresses breast cancer cell invasion.
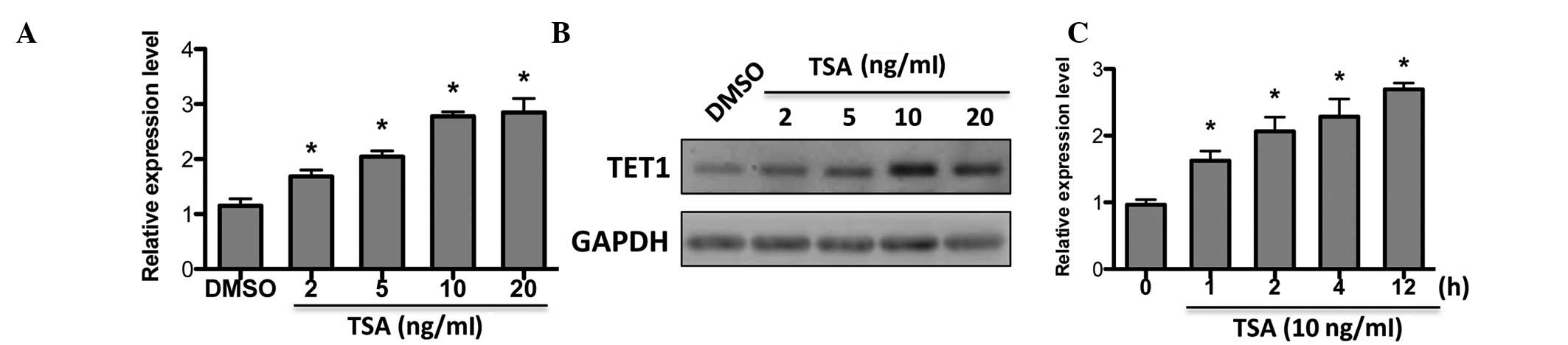
TSA upregulates TET1 expression in MCF-7
cells
Given that the expression of TIMPs and MMPs was
affected by HDAC inhibition and TET1, our hypothesis was that TET1
may be regulated by HDACi. As expected, TET1 expression was
upregulated by TSA in a dose-dependent manner (Fig. 4A), which was confirmed by western
blot analysis (Fig. 4B).
Additionally, TET1 was responsive to short-term TSA treatment
(Fig. 4C). This suggests that TET1
may be a target of TSA in breast cancer.
TET1 knockdown impairs TSA-induced
suppression of breast cancer cell invasion
Since TET1 expression was found to be regulated by
HDAC inhibition, the functional association between TSA and TET1
needed to be elucidated. In order to examine whether TET1 is able
to mediate TSA induced suppression of breast cancer cells, TSA was
subjected to TET1-knockdown and cell invasion was examined. It was
revealed that the inhibitory effect of TSA on breast cancer cell
invasion was impaired in TET1-knockdown cells when compared with
control shRNA expressing cells (Fig.
5A and B). Correspondingly, TSA upregulated TIMP2/3 and
downregulated MMP2/9 expression, which were also impaired in
TET1-knockdown cells. This indicates that TET1 partially mediates
TSA induced repression of breast cancer cell invasion.
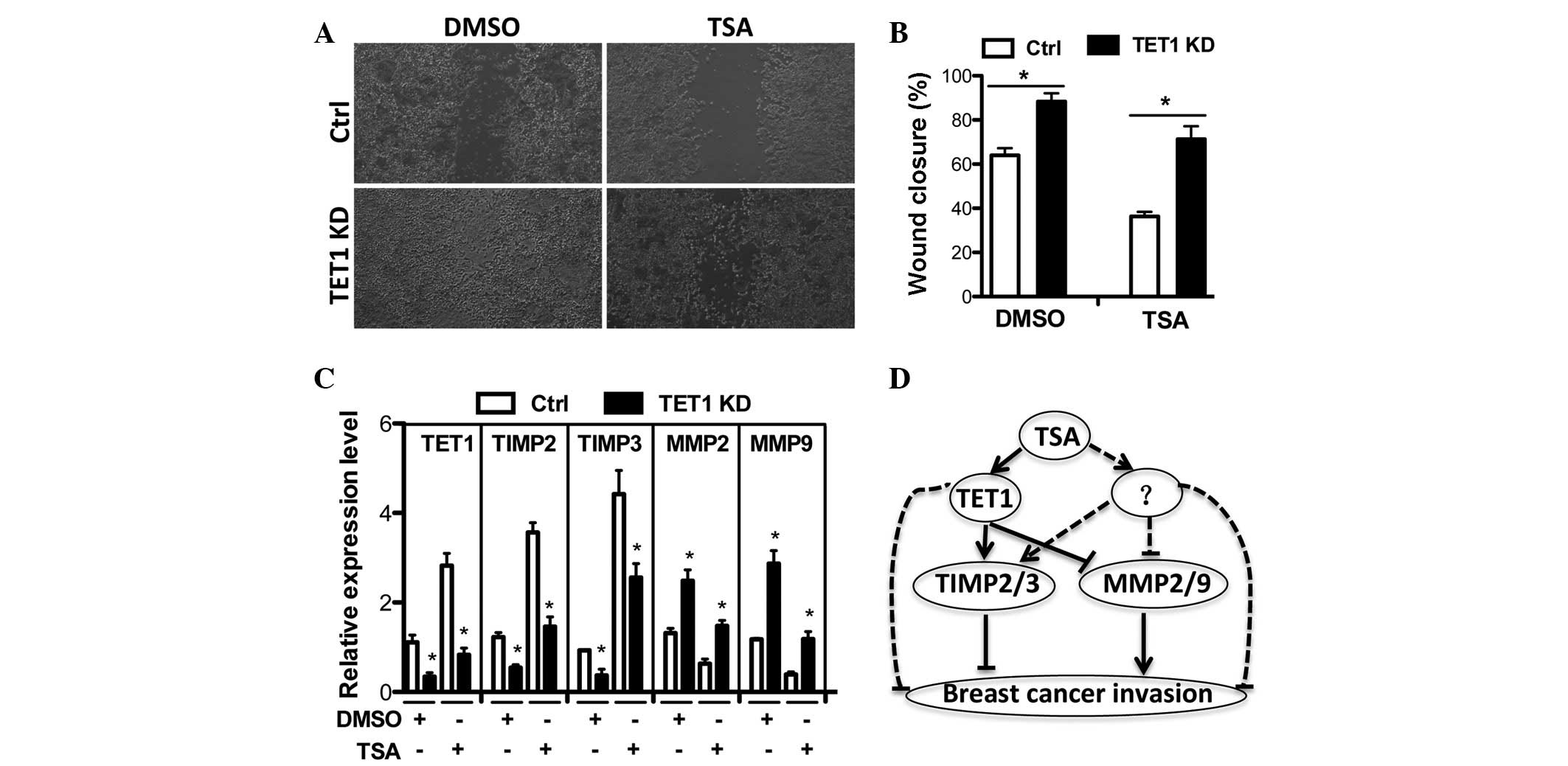 | Figure 5TET1 partially mediates TSA-induced
suppression of breast cancer cell invasion. (A–B) Ctrl or TET1 KD
expressing MCF-7 cells were treated with DMSO or 10 ng/ml of TSA
for 36 h after wounding. (A) Wound closure was monitored and (B)
quantified. (C) Expression levels of TET1, TIMP2, TIMP3, MMP2 and
MMP9 were measured by qRT-PCR in Ctrl or TET1 KD MCF-7 cells under
the condition in (A). (D) Hypothetical model for the action
mechanism of TSA in the suppression of breast cancer cell invasion.
Briefly, TSA regulates TET1 expression or other unknown factors to
promote TIMP2/3 expression and inhibit MMP2/9 transcription leading
to the suppression of breast cancer cell invasion.
*P<0.05 vs. CTRL. TSA, trichostatin A; Ctrl, control
shRNA; KD, knockdown shRNA; qRT-PCR, quantitative real-time PCR;
TIMP, tissue inhibitors of metalloproteinase; MMP, matrix
metalloproteinase. |
Discussion
Breast cancer is one of the most common types of
cancer and has a high risk of mortality among females all over the
world (31). Therefore there is a
great need for understanding the molecular mechanisms underlying
cancer progression and for the development of more effective
therapeutic strategies for breast cancer. Carcinogenesis is able to
be regulated by genetic and epigenetic alterations and epigenetic
alterations are a reversible process, which makes epigenetic
modifications preferable for clinical therapy (32,33).
DNA methylation and histone modifications are the
most important epigenetic mediators of transcriptional regulation
and multiple HDACi are in clinical development for hematological
and solid tumor treatment (34,35).
HDACi result in the accumulation of acetylation in histones and
non-histone proteins to modulate target gene expression in
cancerous cells, which reactivates the expression of tumor
suppressors (35). In the present
study (Fig. 1) and previous
studies (18,36), the non-competitive reversible HDACi
TSA has been demonstrated to suppress breast cancer cell invasion.
The limitations of HDAC inhibition in cancer therapy is the
non-specificity and toxicity of the chemical inhibitors. It is
important to understand the molecular mechanisms in HDACi
treatment. The HDACi valproic acid (VPA) induces ERα expression in
its anti-tumor effects (37) and
TSA enhances the acetylation and stability of the ERα and p300
proteins that may contribute to breast cancer treatment (38). TSA also directly upregulates p53
and RECK, which are important in tumor suppression (21,39).
The present study found that TIMP2/3 were increased
and that MMP2/9 were decreased when treated with TSA (Fig. 1). It has been proposed that TET1
suppresses breast cancer invasion through the activation of TIMPs
and the inhibition of MMPs (22).
Additionally, TET1 and TIMP2/3 reduction was inversely correlated
with MMP2/9 upregulation during breast cancer progression (Fig. 2) (23). Notably, TET1 demonstrated similar
functions in MCF-7 breast cancer cell invasion (Fig. 3) (22). It is reasonable to postulate that
TSA may inhibit breast cancer invasion through the regulation of
TET1 expression. Notably, TSA promoted TET1 expression in a
dose-dependent manner (Fig. 4) and
TET1 partially mediated TSA elicited suppression of cell invasion
(Fig. 5), which was in accordance
with our hypothesis. TET1 expression may be negatively controlled
by a specific HDAC and TSA treatment releases the HDAC inhibition
on TET1 expression. TET1 was found to be associated with TIMPs
genes and promoter regions to regulate their DNA methylation status
and transcription levels (22,40).
Our hypothesis is that TSA may indirectly affect 5-methylcytosine
hydroxylation of TIMP gene promoters through the regulation of TET1
expression, which will be investigated in our future study. There
is a possibility that VPA enhances the global 5-methylcytosine
level in nuclear DNA (41).
Furthermore, the alteration in DNA methylation
status is another important epigenetic modification and DNA
methyltransferases (DNMTs) have become an epigenetic therapy target
in various types of cancer (42).
5-azacytidine, a global DNMT inhibitor, has been approved for
clinical trials against solid tumors (43). Currently, synergistic treatment
with DNMT and HDACi has been applied for producing optimal effects
(41).
As summarized in Fig.
5D, the HDACi TSA may upregulate the expression of TET1, which
results in the activation of TIMPs and the inhibition of MMPs,
thus, leading to the suppression of breast cancer cell invasion.
The present study provides a novel molecular mechanism for HDAC
inhibition in tumor suppression. This may provide insights into the
role of HDACs in cancer development and epigenetic therapy.
Acknowledgements
We would like to thank Shanghai Xuhai Biological
Technology Co., Ltd for the experimental support.
References
|
1
|
Steeg PS: Metastasis suppressors alter the
signal transduction of cancer cells. Nat Rev Cancer. 3:55–63. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mohanam S, Sawaya R, McCutcheon I,
Ali-Osman F, Boyd D and Rao JS: Modulation of in vitro invasion of
human glioblastoma cells by urokinase-type plasminogen activator
receptor antibody. Cancer Res. 53:4143–4147. 1993.PubMed/NCBI
|
|
3
|
Rao JS: Molecular mechanisms of glioma
invasiveness: the role of proteases. Nat Rev Cancer. 3:489–501.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Davies B, Waxman J, Wasan H, Abel P,
Williams G, Krausz T, Neal D, Thomas D, Hanby A and Balkwill F:
Levels of matrix metalloproteases in bladder cancer correlate with
tumor grade and invasion. Cancer Res. 53:5365–5369. 1993.PubMed/NCBI
|
|
5
|
Horikoshi M: Histone acetylation: from
code to web and router via intrinsically disordered regions. Curr
Pharm Des. 19:5019–5042. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gräff J and Tsai LH: Histone acetylation:
molecular mnemonics on the chromatin. Nat Rev Neurosci. 14:97–111.
2013.PubMed/NCBI
|
|
7
|
Mayo MW, Denlinger CE, Broad RM, Yeung F,
Reilly ET, Shi Y and Jones DR: Ineffectiveness of histone
deacetylase inhibitors to induce apoptosis involves the
transcriptional activation of NF-kappa B through the Akt pathway. J
Biol Chem. 278:18980–18989. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brochier C, Dennis G, Rivieccio MA,
McLaughlin K, Coppola G, Ratan RR and Langley B: Specific
acetylation of p53 by HDAC inhibition prevents DNA damage-induced
apoptosis in neurons. J Neurosci. 33:8621–8632. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sachweh MC, Drummond CJ, Higgins M,
Campbell J and Laín S: Incompatible effects of p53 and HDAC
inhibition on p21 expression and cell cycle progression. Cell Death
Dis. 4:e5332013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cheung WL, Briggs SD and Allis CD:
Acetylation and chromosomal functions. Curr Opin Cell Biol.
12:326–333. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Van Lint C, Emiliani S and Verdin E: The
expression of a small fraction of cellular genes is changed in
response to histone hyperacetylation. Gene Expr. 5:245–253.
1996.PubMed/NCBI
|
|
12
|
Mitsiades CS, Mitsiades NS, McMullan CJ,
et al: Transcriptional signature of histone deacetylase inhibition
in multiple myeloma: biological and clinical implications. Proc
Natl Acad Sci USA. 101:540–545. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Song J, Noh JH, Lee JH, et al: Increased
expression of histone deacetylase 2 is found in human gastric
cancer. APMIS. 113:264–268. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shao Y, Gao Z, Marks PA and Jiang X:
Apoptotic and autophagic cell death induced by histone deacetylase
inhibitors. Proc Natl Acad Sci USA. 101:18030–18035. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Marks PA, Richon VM, Miller T and Kelly
WK: Histone deacetylase inhibitors. Adv Cancer Res. 91:137–168.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
McGarry LC, Winnie JN and Ozanne BW:
Invasion of v-Fos(FBR)-transformed cells is dependent upon histone
deacetylase activity and suppression of histone deacetylase
regulated genes. Oncogene. 23:5284–5292. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marks PA, Richon VM, Kelly WK, Chiao JH
and Miller T: Histone deacetylase inhibitors: development as cancer
therapy. Novartis Found Symp. 259:269–281. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vigushin DM, Ali S, Pace PE, et al:
Trichostatin A is a histone deacetylase inhibitor with potent
antitumor activity against breast cancer in vivo. Clin Cancer Res.
7:971–976. 2001.
|
|
19
|
Tarasenko N, Nudelman A, Tarasenko I, et
al: Histone deacetylase inhibitors: the anticancer, antimetastatic
and antiangiogenic activities of AN-7 are superior to those of the
clinically tested AN-9 (Pivanex). Clin Exp Metastasis. 25:703–716.
2008. View Article : Google Scholar
|
|
20
|
Bolden JE, Peart MJ and Johnstone RW:
Anticancer activities of histone deacetylase inhibitors. Nat Rev
Drug Discov. 5:769–784. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu LT, Chang HC, Chiang LC and Hung WC:
Histone deacetylase inhibitor up-regulates RECK to inhibit MMP-2
activation and cancer cell invasion. Cancer Res. 63:3069–3072.
2003.PubMed/NCBI
|
|
22
|
Hsu CH, Peng KL, Kang ML, et al: TET1
suppresses cancer invasion by activating the tissue inhibitors of
metalloproteinases. Cell Rep. 2:568–579. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang H, Liu Y, Bai F, et al: Tumor
development is associated with decrease of TET gene expression and
5-methylcytosine hydroxylation. Oncogene. 32:663–669. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Asrani K, Keri RA, Galisteo R, et al: The
HER2- and heregulin beta1 (HRG)-inducible TNFR superfamily member
Fn14 promotes HRG-driven breast cancer cell migration, invasion,
and MMP9 expression. Mol Cancer Res. 11:393–404. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Brown SA, Ghosh A and Winkles JA:
Full-length, membrane-anchored TWEAK can function as a juxtacrine
signaling molecule and activate the NF-kappaB pathway. J Biol Chem.
285:17432–17441. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Grimaldi C, Pisanti S, Laezza C, et al:
Anandamide inhibits adhesion and migration of breast cancer cells.
Exp Cell Res. 312:363–373. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Albini A, Melchiori A, Santi L, Liotta LA,
Brown PD and Stetler-Stevenson WG: Tumor cell invasion inhibited by
TIMP-2. J Natl Cancer Inst. 83:775–779. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Baker AH, George SJ, Zaltsman AB, Murphy G
and Newby AC: Inhibition of invasion and induction of apoptotic
cell death of cancer cell lines by overexpression of TIMP-3. Br J
Cancer. 79:1347–1355. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jinga D, Stefanescu M, Blidaru A, Condrea
I, Pistol G and Matache C: Serum levels of matrix
metalloproteinases MMP-2 and MMP-9 and their tissue natural
inhibitors in breast tumors. Roum Arch Microbiol Immunol.
63:141–158. 2004.PubMed/NCBI
|
|
30
|
Sier CF, Kubben FJ, Ganesh S, et al:
Tissue levels of matrix metalloproteinases MMP-2 and MMP-9 are
related to the overall survival of patients with gastric carcinoma.
Br J Cancer. 74:413–417. 1996. View Article : Google Scholar
|
|
31
|
Jemal A, Siegel R, Ward E, et al: Cancer
statistics, 2008. CA Cancer J Clin. 58:71–96. 2008. View Article : Google Scholar
|
|
32
|
Taby R and Issa JP: Cancer epigenetics. CA
Cancer J Clin. 60:376–392. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cai FF, Kohler C, Zhang B, Wang MH, Chen
WJ and Zhong XY: Epigenetic therapy for breast cancer. Int J Mol
Sci. 12:4465–4487. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Namdar M, Perez G, Ngo L and Marks PA:
Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA
damage and sensitizes transformed cells to anticancer agents. Proc
Natl Acad Sci USA. 107:20003–20008. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu T, Kuljaca S, Tee A and Marshall GM:
Histone deacetylase inhibitors: multifunctional anticancer agents.
Cancer Treat Rev. 32:157–165. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang X, Phillips DL, Ferguson AT, Nelson
WG, Herman JG and Davidson NE: Synergistic activation of functional
estrogen receptor (ER)-alpha by DNA methyltransferase and histone
deacetylase inhibition in human ER-alpha-negative breast cancer
cells. Cancer Res. 61:7025–7029. 2001.
|
|
37
|
Travaglini L, Vian L, Billi M, Grignani F
and Nervi C: Epigenetic reprogramming of breast cancer cells by
valproic acid occurs regardless of estrogen receptor status. Int J
Biochem Cell Biol. 41:225–234. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kim SH, Kang HJ, Na H and Lee MO:
Trichostatin A enhances acetylation as well as protein stability of
ERalpha through induction of p300 protein. Breast Cancer Res.
12:R222010. View Article : Google Scholar
|
|
39
|
Henderson C, Mizzau M, Paroni G, Maestro
R, Schneider C and Brancolini C: Role of caspases, Bid, and p53 in
the apoptotic response triggered by histone deacetylase inhibitors
trichostatin-A (TSA) and suberoylanilide hydroxamic acid (SAHA). J
Biol Chem. 278:12579–12589. 2003. View Article : Google Scholar
|
|
40
|
Williams K, Christensen J, Pedersen MT, et
al: TET1 and hydroxymethylcytosine in transcription and DNA
methylation fidelity. Nature. 473:343–348. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kristensen LS, Nielsen HM and Hansen LL:
Epigenetics and cancer treatment. Eur J Pharmacol. 625:131–142.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hatziapostolou M and Iliopoulos D:
Epigenetic aberrations during oncogenesis. Cell Mol Life Sci.
68:1681–1702. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chik F and Szyf M: Effects of specific
DNMT gene depletion on cancer cell transformation and breast cancer
cell invasion; toward selective DNMT inhibitors. Carcinogenesis.
32:224–232. 2011. View Article : Google Scholar : PubMed/NCBI
|