Introduction
The human ubiquitin-specific protease 22
(USP22) gene is located on chromosome 17p11.2. Its product
is a 525 amino acid protein with a molecular weight of ~60 kDa that
functions as a deubiquitinating enzyme in vivo and in
vitro (1). In humans, USP22 is
a subunit of the SAGA coactivator complex that is required for
activator-driven transcription. Substrates of USP22 include the
histones H2A and H2B, the shelterin protein TRF1, the transcription
factor p53, and FBP1 (2–5). In addition, USP22 is overexpressed in
numerous human tumors and is associated with treatment resistance,
tumor aggressiveness and increased metastasis in cancer patients
(6,7). However, little is known regarding the
transcriptional regulation of USP22.
Acetylation is a posttranslational modification that
regulates the physiological functions of proteins. Histone
acetylases (HAT) and deacetylases (HDACs) regulate the dynamic
equilibrium of acetylation (8).
HDAC inhibitors have been identified as a novel class of potential
drugs for use in cancer therapy, based on observations that the
overexpression of HDAC was associated with tumorigenesis (9). The antitumor effects of HDAC
inhibitors are due to the transcriptional alteration of specific
cancer-related genes, including regulators of the cell cycle,
apoptosis, differentiation and invasion (10–12).
Therefore, it is hypothesized that HDAC-induced apoptosis may alter
USP22 expression.
The aim of the present study was to determine the
effect of the HDAC inhibitor trichostatin A (TSA) on the expression
levels of endogenous USP22 and its promoter activity in human
cancer cells. In addition, the mechanisms underlying the anticancer
activity of TSA were investigated.
Materials and methods
Cell culture
Human HeLa cells (American Type Culture Collection,
Manassas, VA, USA) were used in this study. The cells were cultured
in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen Life
Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine
serum (Gibco-BRL, Carlsbad, CA, USA) at 37°C with 5%
CO2.
Total RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA extraction and qPCR were performed as
described previously (13).
Briefly, total RNA was isolated from cells using TRIzol Reagent
(Invitrogen), and reverse-transcribed using M-MLV reverse
transcriptase. Real-time PCR was performed using SYBR Green PCR
Master Mix (Takara Bio, Dalian, China) on an ABI 7500 Real-Time PCR
System (Applied Biosystems, Carlsbad, CA, USA). The mRNA levels of
USP22 and GAPDH were measured with the following
specific primers: Forward: 5′-GTGTCTTCTTCGGCTGTTTA-3′ and reverse:
5′-CTCCTCCTTGGCGATTATTT-3′ for USP22; and forward:
5′-AGAAGGCTGGGGCTCATTTG-3′ and reverse: 5′-AGGGGCCATCCACAGTCTTC-3′
for GAPDH. All qPCR results are presented as the mean of at
least three independent experiments using duplicate qPCR
analysis.
Western blotting
Whole cell lysates were separated by SDS-PAGE using
10% polyacrylamide gels and transferred to polyvinylidene fluoride
membranes (Bio-Rad, Hercules, CA, USA). After overnight blocking at
4°C in a buffer containing 5% (wt/vol) non-fat milk powder, the
membrane was incubated in fresh buffer with the appropriate
antibody for 1 h at room temperature. Monoclonal antibodies against
USP22 and GAPDH were purchased from Santa Cruz Biotechnology, Inc.
(sc-69082 and sc-20358; Dallas, TX, USA). The antigen-antibody
complex was detected by incubating the membrane for 1 h at room
temperature in buffer containing a 1:10,000 dilution of horseradish
peroxidase-conjugated anti-goat IgG secondary antibody (Santa Cruz
Biotechnology) Western blots were visualized using an enhanced
chemiluminescence system (Pierce Biotechnology, Inc. Rockford, IL,
USA).
Plasmid constructs
The USP22 promoters were constructed as described
previously (13). To construct
CMV-driven USP22 expression vectors, total RNA was isolated from
human HeLa cells and reverse-transcribed using the reverse
transcriptase M-MLV primed by an oligo (dT)15 primer. The primers
used in the subsequent PCR amplification of USP22 cDNA were as
follows: Forward: 5′-GAAGCTTATGAGCGACCAAGATCACTCCATG-3′ and
reverse: 5′-GGAATTCTCAGAAGCCATTGCCACTGA-3′. The PCR products were
analyzed on agarose gels and digested with HindIII and
EcoRI and ligated into pCMV-His with T4 DNA ligase (Takara
Bio). The fidelity of the constructs was confirmed by sequencing.
The sequencing primer was TAATACGACTCACTATAGGG.
Luciferase assay
Cells were plated onto 24-well plates at a density
of 5×104 cells per well and left overnight. This was
followed by transfection with promoter reporter plasmid DNA using
Lipofectamine 2000 reagent (Invitrogen Life Technologies). The
media were changed 24 h later, for medium with or without TSA, and
cell lysates were collected at the times indicated for analysis by
luciferase assay using a Dual Luciferase Assay kit (Promega,
Madison, WI, USA). All transfections were performed in
triplicate.
Chromatin immunoprecipitation (ChIP)
ChIP was performed as described previously (13). Briefly, cells were fixed by adding
formaldehyde to a final concentration of 1% and incubated with
moderate shaking for 30 min at room temperature. Thereafter, cells
were washed twice with cold phosphate-buffered saline (PBS). The
pellet was resuspended and lysed, and nuclei were isolated and
sonicated until the chromatin had an average length of 500–1500 bp.
Chromatin fragments were immunoprecipitated from cells using rabbit
polyclonal antibody against RNA polymerase II (sc-899x; Santa Cruz
Biotechnology) DNA samples from the immunoprecipitates and control
inputs were analyzed using qPCR with the following primers:
forward, 5′-GTCTACCCAGAGCCTAACGG-3′, and reverse
5′-GCGGAGGCCGGACAAAGATGGG-3′.
Flow cytometry
Cells (1×105) were seeded onto 6-well
plates, incubated overnight and transfected with USP22 or an empty
vector. The media were changed 24 h later for media containing TSA,
and cells were incubated for an additional 24 or 48 h. The cells
were then harvested using trypsin, washed with 1X PBS, and
resuspended in 50 μl PBS. Cell death was assessed by flow cytometry
(FACSCalibur; BD Biosciences, Franklin Lakes, NJ, USA) following
staining with fluorescein isothiocyanate-Annexin V and potassium
iodide.
Statistical analysis
Each experiment was performed at least three times
independently. The SPSS 13.0 software program (SPSS, Inc., Chicago,
IL, USA) was used for statistical analysis. The data between two
groups was analyzed by a Student’s t-test. The data between
multiple groups were analyzed by one-way analysis of variance.
P<0.05 was considered to indicate a statistically significant
difference.
Results
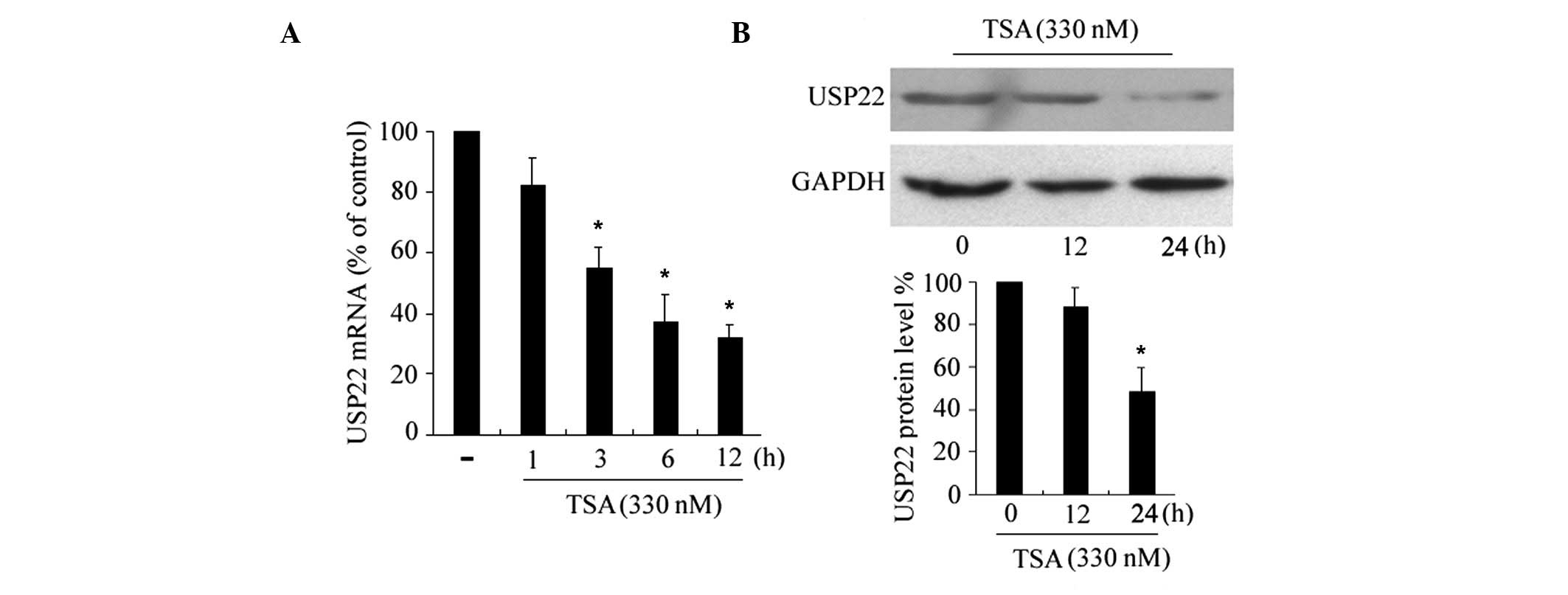
TSA suppresses USP22 expression
To assess the effects of HDAC inhibitors on USP22
expression, HeLa cells were incubated with apoptosis-inducing
concentrations of TSA (330 nM) (14) for different time periods, and
USP22 mRNA levels were examined by RT-qPCR. As shown in
Fig. 1A, the USP22 mRNA
expression levels were reduced marginally following a 3 h
incubation with TSA and were reduced significantly after 6–12 h,
indicating that the suppression is a time-dependent. The effects of
TSA on the expression levels of the USP22 protein were determined
by western blotting. Consistent with the reduction in USP22
mRNA, TSA downregulated the expression levels of USP22 protein
(Fig. 1B), with a significant
reduction following 24 h of treatment. In addition, TSA
downregulated the expression of USP22 in another cancer cell line,
HepG2 (data not shown).
To exclude the possibility that TSA affected the
stability of USP22 mRNA, actinomycin D was used to block
mRNA transcription, and the levels of USP22 mRNA were
assessed with RT-qPCR. When cells were maintained in normal culture
conditions, the half-life of USP22 mRNA was ~6 h following
the inhibition of transcription. When cells were cotreated with TSA
and actinomycin D the half-life of USP22 mRNA was unchanged,
indicating that TSA does not affect the stability of USP22
mRNA.
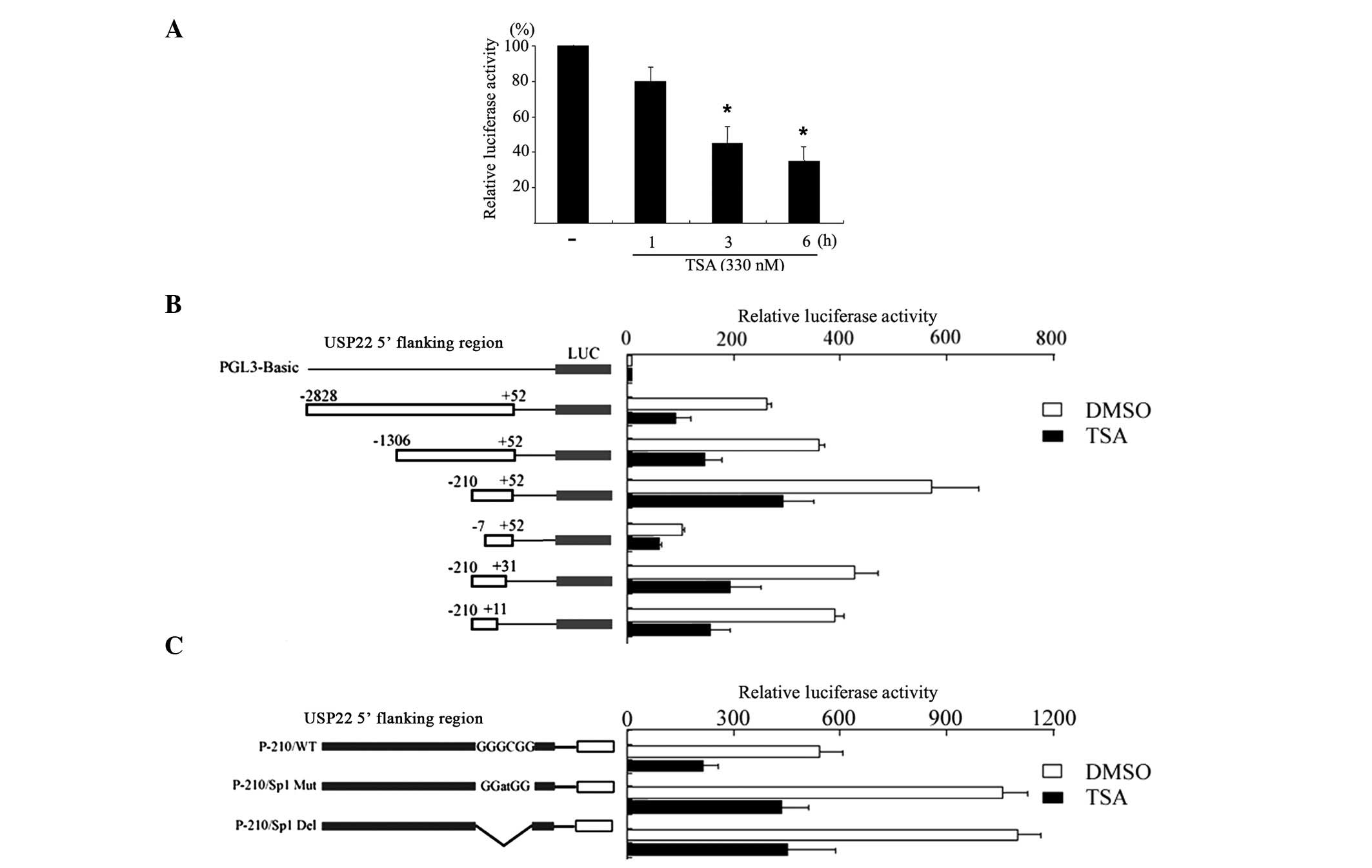
TSA suppresses USP22 promoter
activity
Subsequently, luciferase assays were used to assess
the effects of TSA on the activity of the USP22 promoter.
The 2.8 kb wild-type USP22 promoter P-2828/+52 was transfected into
HeLa cells and incubated for 24 h. Following incubation, cells were
treated with TSA for different time periods. As shown in Fig. 2A, treatment with TSA reduced the
promoter activity in a time-dependent manner, which is consistent
with the reduction of the USP22 mRNA expression levels.
Subsequently, a series of truncated promoter-reporter gene
constructs, derived from p-2828/+52, were transfected into cells to
characterize the regulatory sequence that responds to TSA. Notably,
the promoter activity of the 5′ deletion constructs p-2828/+52,
p-1306/+52, p-210/+52, and p-7/+52 and the 3′ deletion constructs
p-210/+31, p-210/+11 were all reduced by treatment with TSA
compared with that of the untreated cells (Fig. 2B). Finally, the activity of a
promoter harboring a mutated Sp1 binding site (p-210/sp1mut)
located between -7 and -13 was also reduced by 40% following
treatment with TSA compared with that of the wild-type promoter
p-210/+52 (Fig. 2C). This suggests
that the Sp1 binding site is not involved in the TSA-mediated
suppression of USP22 promoter activity.
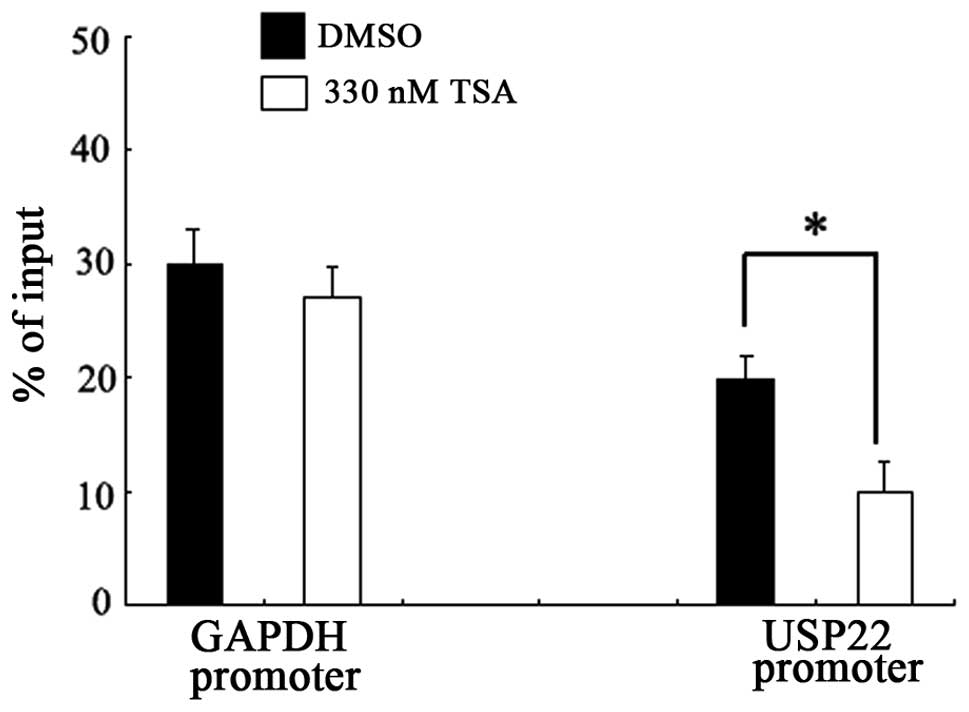
TSA-induced downregulation of USP22
involves the recruitment of RNA polymerase II
Recent studies have demonstrated that HDAC activity
is required for the recruitment of RNA polymerase II to several
specific gene promoters (15).
Since no regulatory elements were characterized that were
responsive to TSA repression up or downstream of the USP22
gene transcription start site, the effect of TSA on the recruitment
of RNA polymerase II to the USP22 promoter was investigated
using a ChIP assay. As shown in Fig.
3, TSA treatment significantly blocked the binding of RNA
polymerase II to the USP22 promoter in HeLa cells. This
inhibitory effect was not detected with the control promoter region
of GAPDH, indicating that the inhibition was specific to the
USP22 promoter.
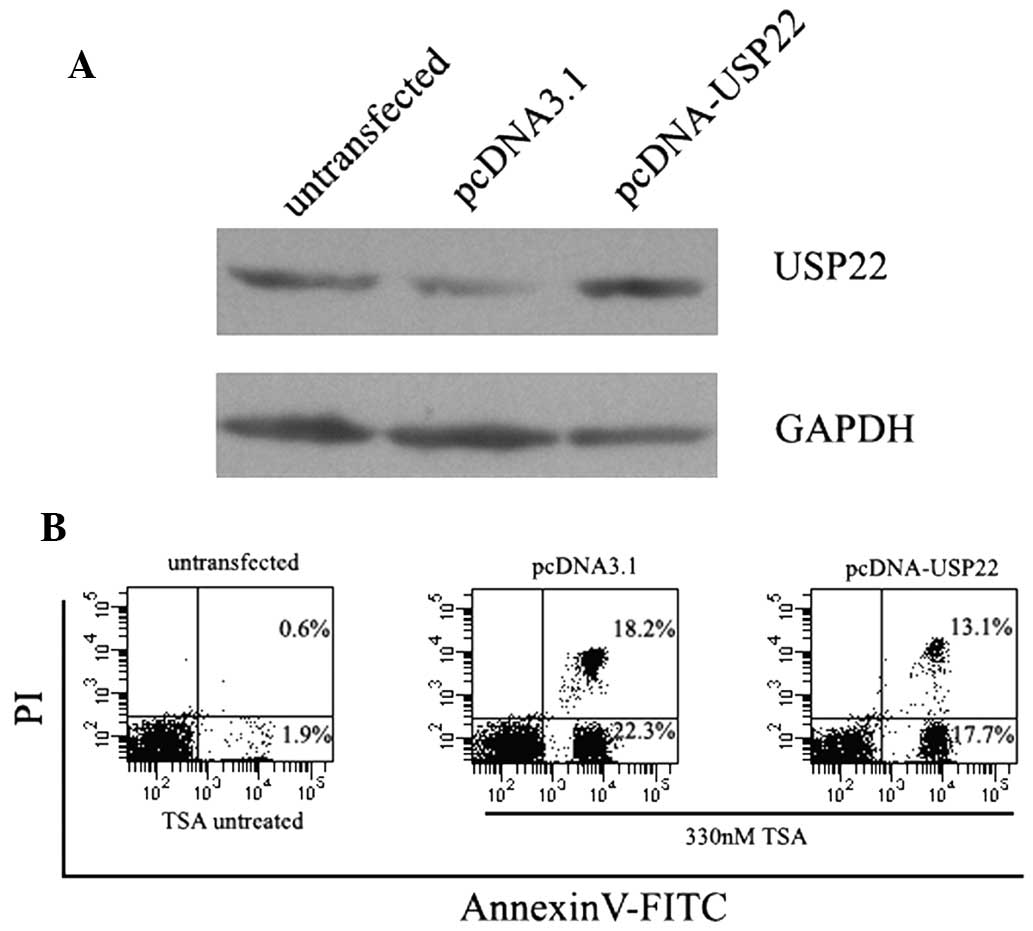
Overexpression of USP22 attenuates
TSA-induced apoptosis
The effect of USP22 overexpression on TSA-induced
apoptosis was investigated. A CMV-driven USP22 expression vector
was generated and used to transiently transfect HeLa cells. As
shown in Fig. 4, a significant
increase in the expression levels of USP22 protein was observed in
pcDNA-USP22 transfected cells, whereas no change was detected in
empty vector-transfected cells. As expected, the overexpression of
USP22 significantly decreased TSA-induced apoptosis in HeLa cells
(Fig. 4).
Discussion
Recently, research interest in the expression of
USP22 has increased. In embryogenesis, USP22 is expressed
periodically and its depletion may lead to early embryonic
mortality. Furthermore, the overexpression of USP22 has been
observed in the majority of types of human cancer cell and has been
linked to cancer progression (2,6).
Therefore, the reduction of USP22 expression levels may provide a
novel cancer therapy. However, the mechanisms that regulate USP22
expression, particularly in human tumor cells remain to be
elucidated. The results of the current study revealed for the first
time that a HDAC inhibitor negatively regulates USP22
expression.
TSA is a classic HDAC inhibitor that induces
apoptosis by altering the expression levels of pro- and
anti-apoptotic genes. For example, the expression levels of the
cyclin-dependent kinase inhibitor p21 were upregulated during
TSA-induced apoptosis (12),
whereas the expression levels of survivin, an apoptosis inhibitor,
were reduced (11). As USP22 is an
anti-apoptotic protein, it was hypothesized that USP22 is
downregulated by TSA, a hypothesis that was supported by the
results of the qPCR and western blot analysis. However, the
mechanisms that regulate TSA-induced gene silencing are complex,
and not completely understood. Certain studies have indicated that
TSA reduces gene expression at the transcriptional level (10), whereas others reported that TSA
suppresses gene expression by affecting mRNA stability (16). An additional study revealed that
TSA may lead to protein degradation (17). In the present study, TSA inhibited
USP22 expression at the transcription level, and did not affect the
half-life of USP22 mRNA. The TSA-mediated suppression of
USP22 was an early, time-dependent event. These results are
supported by a previous study that determined that USP22
transcription is regulated by extracellular signals (18).
Since TSA suppressed the expression levels of
USP22 mRNA, one of the aims of the current study was to
characterize the specific elements in the USP22 promoter
that were targeted by TSA. Although several regulatory elements
were found in the USP22 promoter, no DNA element up- or
downstream of the transcription start site was responsive to TSA,
including the Sp1 binding site that was identified close to the
USP22 transcription start site. A previous study suggested that Sp1
is important in HDAC inhibitor-induced apoptosis (19). Sp1 is a transcription factor that
is acetylated in the DNA binding domain by CBP/p300 and
subsequently modulates transcriptional activity and influences
protein-protein interactions (20). Therefore, it was originally
hypothesized that TSA-stimulated suppression of USP22 expression
was mediated by this site. However, promoter deletion and
mutagenesis analyses showed that the Sp1 binding site was not
involved in TSA-induced USP22 downregulation.
TSA responsive regions were not identified up- or
downstream of the USP22 transcription start site, indicating
that the key region that mediates TSA-induced USP22
suppression may be located close to the transcription initiation
site. A previous study suggested that histone deacetylase activity
is required to recruit RNA polymerase II to promoters, thus
affecting gene transcription (15). Therefore, interfering with RNA
polymerase recruitment is one mechanism by which HDAC inhibitors
stimulate gene silencing (21). In
the present study, ChIP analysis revealed that TSA blocked the
recruitment of RNA polymerase II, indicating that TSA blocks the
formation of the preinitiation complex at the USP22
promoter. Nevertheless, the mechanism by which HDAC activity
affected the binding of RNA polymerase II to the USP22
promoter requires further investigation.
To confirm the anti-apoptotic role of TSA treatment,
a USP22 expression vector driven by a CMV promoter was produced.
Since the activity of the CMV promoter increases marginally in
response to TSA treatment (22),
it was possible to obtain USP22 over-expressing HeLa cells
following treatment with TSA. The overexpression of USP22
attenuates TSA-induced apoptosis, indicating that USP22 has an
important role in HDAC inhibitor-induced apoptotic signaling.
Although the mechanism by which USP22 attenuates TSA-induced
apoptosis is not yet fully understood, the regulation of
apoptosis-related genes by USP22 is a possible route. Indeed,
several key genes in TSA-induced apoptosis, such as the
cyclin-dependent kinase inhibitor p21 WAF1, are also modulated by
USP22. p21 is a downstream target gene of USP22, and
downregulation of USP22 promoted p21 expression. By contrast, p21
is upregulated during TSA-induced apoptosis. Therefore, the
suppression of USP22 expression by TSA may regulate p21 expression.
In addition, p53 may also participate in the TSA-USP22 pathway as
it has been linked to TSA-induced apoptosis. Whilst USP22-mediated
stabilization of SIRT1 has been shown to lead to a reduction in the
levels of p53 acetylation and the suppression of p53-mediated
apoptosis (4), contradictory
studies have reported that the activity and stability of SIRT1 are
not affected by USP22 (23). These
contrasting results could be due to cell-specific effects. A
greater understanding of the mechanisms by which USP22 regulates
gene transcription may identify additional molecules that are
involved in TSA-induced apoptosis.
In conclusion, the present study revealed that the
expression of USP22 is negatively regulated by TSA. Furthermore, it
was determined that this negative regulation occurred via
interference of the binding between RNA polymerase II and the
USP22 promoter. In addition, the exogenous overexpression of
USP22 attenuated TSA-induced apoptosis. These results indicate that
blocking USP22 expression at the transcriptional level by TSA may
provide a novel strategy for cancer therapy.
Acknowledgements
This study was supported by the National Nature
Science Foundation of China (grant no. 31000581) and the Jiangxi
Science and Technology Support Programmer, P.R. China (grant no.
2010BSA14000).
References
|
1
|
Lee HJ, Kim MS, Shin JM, Park TJ, Chung HM
and Baek KH: The expression patterns of deubiquitinating enzymes,
USP22 and Usp22. Gene Expr Patterns. 6:277–284. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang XY, Varthi M, Sykes SM, et al: The
putative cancer stem cell marker USP22 is a subunit of the human
SAGA complex required for activated transcription and cell-cycle
progression. Mol Cell. 29:102–111. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhao Y, Lang G, Ito S, et al: A TFTC/STAGA
module mediates histone H2A and H2B deubiquitination, coactivates
nuclear receptors, and counteracts heterochromatin silencing. Mol
Cell. 29:92–101. 2008. View Article : Google Scholar
|
|
4
|
Lin Z, Yang H, Kong Q, et al: USP22
antagonizes p53 transcriptional activation by deubiquitinating
Sirt1 to suppress cell apoptosis and is required for mouse
embryonic development. Mol Cell. 46:484–494. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Atanassov BS and Dent SY: USP22 regulates
cell proliferation by deubiquitinating the transcriptional
regulator FBP1. EMBO Rep. 12:924–930. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Glinsky GV: Death-from-cancer signatures
and stem cell contribution to metastatic cancer. Cell Cycle.
4:1171–1175. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Glinsky GV, Berezovska O and Glinskii AB:
Microarray analysis identifies a death-from-cancer signature
predicting therapy failure in patients with multiple types of
cancer. J Clin Invest. 115:1503–1521. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kouzarides T: Acetylation: a regulatory
modification to rival phosphorylation? EMBO J. 19:1176–1179. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kawai H, Li H, Avraham S, Jiang S and
Avraham HK: Overexpression of histone deacetylase HDAC1 modulates
breast cancer progression by negative regulation of estrogen
receptor alpha. Int J Cancer. 107:353–358. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Duan H, Heckman CA and Boxer LM: Histone
deacetylase inhibitors down-regulate bcl-2 expression and induce
apoptosis in t(14;18) lymphomas. Mol Cell Biol. 25:1608–1619. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hsu YF, Sheu JR, Lin CH, et al:
Trichostatin A and sirtinol suppressed survivin expression through
AMPK and p38MAPK in HT29 colon cancer cells. Biochim Biophys Acta.
1820:104–115. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Han JW, Ahn SH, Kim YK, et al: Activation
of p21(WAF1/Cip1) transcription through Sp1 sites by histone
deacetylase inhibitor apicidin: involvement of protein kinase C. J
Biol Chem. 276:42084–42090. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xiong J, Che X, Li X, Yu H, Gong Z and Li
W: Cloning and characterization of the human USP22 gene promoter.
PLoS One. 7:e527162012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sakamoto S, Potla R and Larner AC: Histone
deacetylase activity is required to recruit RNA polymerase II to
the promoters of selected interferon-stimulated early response
genes. J Biol Chem. 279:40362–40367. 2004. View Article : Google Scholar
|
|
15
|
Zhang Y and Dufau ML: Silencing of
transcription of the human luteinizing hormone receptor gene by
histone deacetylase-mSin3A complex. J Biol Chem. 277:33431–33438.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xiong Y, Dowdy SC, Podratz KC, et al:
Histone deacetylase inhibitors decrease DNA methyltransferase-3B
messenger RNA stability and down-regulate de novo DNA
methyltransferase activity in human endometrial cells. Cancer Res.
65:2684–2689. 2005. View Article : Google Scholar
|
|
17
|
Chen WY, Weng JH, Huang CC and Chung BC:
Histone deacetylase inhibitors reduce steroidogenesis through
SCF-mediated ubiquitination and degradation of steroidogenic factor
1 (NR5A1). Mol Cell Biol. 27:7284–7290. 2007. View Article : Google Scholar
|
|
18
|
Ovaa H, Kessler BM, Rolen U, Galardy PJ,
Ploegh HL and Masucci MG: Activity-based ubiquitin-specific
protease (USP) profiling of virus-infected and malignant human
cells. Proc Natl Acad Sci USA. 101:2253–2258. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Waby JS, Chirakkal H, Yu C, et al: Sp1
acetylation is associated with loss of DNA binding at promoters
associated with cell cycle arrest and cell death in a colon cell
line. Mol Cancer. 9:2752010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Suzuki T, Kimura A, Nagai R and Horikoshi
M: Regulation of interaction of the acetyltransferase region of
p300 and the DNA-binding domain of Sp1 on and through DNA binding.
Genes Cells. 5:29–41. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Furumai R, Ito A, Ogawa K, et al: Histone
deacetylase inhibitors block nuclear factor-kappaB-dependent
transcription by interfering with RNA polymerase II recruitment.
Cancer Sci. 102:1081–1087. 2011. View Article : Google Scholar
|
|
22
|
Choi KH, Basma H, Singh J and Cheng PW:
Activation of CMV promoter-controlled glycosyltransferase and
beta-galactosidase glycogenes by butyrate, tricostatin A, and
5-aza-2′-deoxycytidine. Glycoconj J. 22:63–69. 2005.PubMed/NCBI
|
|
23
|
Armour SM, Bennett EJ, Braun CR, et al: A
high-confidence interaction map identifies SIRT1 as a mediator of
acetylation of USP22 and the SAGA coactivator complex. Mol Cell
Biol. 33:1487–1502. 2013. View Article : Google Scholar
|