Introduction
Ovarian cancer is one of the most common
gynecological malignancies. Currently, debulking surgery and
cisplatin-based chemotherapy are the recommended approaches with
the highest curative potential (1). However, acquired chemoresistance is a
major obstacle that affects the success rate of ovarian cancer
treatment. The five-year survival rate for ovarian cancer patients
is currently ~30% (2). Previous
studies have researched the molecular alterations in
cisplatin-resistant cancer cells, however, the underlying
mechanisms promoting cisplatin resistance in ovarian cancer cells
remain to be elucidated (3).
Previous studies have suggested that autophagy may
have a role in cancer cell chemoresistance (4,5).
Autophagy is a cellular process of self destruction which occurs in
all eukaryotic cells. Damaged organelles and molecules are engulfed
by autophagosomes and degraded by lysosomal hydrolases, for energy
recycling (6). Dysregulation of
autophagy has been associated with numerous diseases, including
cancer (7); however, the role of
autophagy in cancer chemoresistance remains unknown.
Inhibition of autophagy by 3-methyladenine (3-MA),
or Beclin 1 small interfering (si)RNA, was previously shown to
increase chemotherapy-induced apoptosis in human hepatocellular
carcinoma cells, and inhibit tumor growth in a mouse xenograft
model (8). Furthermore, knockdown
of Beclin 1 and autophagy-related protein 7 (ATG7) expression
levels, in OE19 and KYSE450 esophageal cancer cells, enhanced the
effects of 5-fluorouracil (5-FU), a chemotherapeutic agent used to
treat esophageal cancer (9). These
observations suggest that inhibitors of autophagy may be potential
targets to improve the therapeutic efficacy of conventional
chemotherapeutics. Conversely, in MCF-7 breast cancer cells,
knockdown of migration inhibitory factor expression, was shown to
enhance the cytotoxicity of doxorubicin and etoposide, by inducing
autophagy (10). A previous study
using H460/cis cisplatin-resistant lung carcinoma cells
additionally showed that the levels of microtubule-associated
protein 1 light chain 3 (LC3), and autophagosome formation were
significantly lower in resistant cells, as compared with their
non-resistant parental cells (11). Furthermore, a co-treatment of
cisplatin with trifluoperazine, an inducer of autophagy, sensitized
H460/cis cells to cisplatin, suggesting that the decreased levels
of autophagy may promote cisplatin resistance in lung cancer.
Currently, no consistent conclusions have been made regarding the
role of autophagy in chemoresistance. Therefore, the aim of the
present study was to investigate the role of autophagy in mediating
cisplatin resistance in human ovarian cancer cells, and explore
autophagy as a potential target for ovarian cancer treatment.
Materials and methods
Cell lines and reagents
The A2780 cisplatin-sensitive human ovarian cancer
cell line, and the A2780cp cisplatin-resistant clone, were obtained
from the Shanghai Key Laboratory of Female Reproductive Endocrine
Related Diseases (Shanghai, China). The A2780 cisplatin-sensitive
cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM;
Gibco Life Technologies, Carlsbad, CA, USA), supplemented with 10%
fetal bovine serum (FBS South American origin; Bio-west, Logan, UT,
USA), 100 U/ml penicillin and 100 μg/ml streptomycin
(Sigma-Aldrich, St.Louis, MO, USA), in a humidified 5%
CO2 atmosphere, at 37°C. A2780cp cells were grown in
DMEM, supplemented with 10% FBS and 1 μg/ml cisplatin, to maintain
resistance. The cisplatin was purchased from Hansoh Pharmaceutical
Co., Ltd. (Lianyungang, Jiangsu, China), and the 3-MA (M9281;
Sigma-Aldrich) was dissolved in sterile double distilled water at
65°C.
Cell viability assay
The A2780 and A2780cp cells were seeded at
1×104 cells/well, in 96-well plates. The following day,
various concentrations (5–50 μg/ml) of cisplatin were added to the
wells and the plates were incubated for 24 h. Each treatment was
applied to four wells. The cell viability was assessed using a
water soluble tetrazolium salt-8 (WST-8) Cell Counting kit (Dojindo
Molecular Technologies, Inc., Kumamoto, Japan). Briefly, 10 μl
WST-8 and 100 μl DMEM was added to each well and incubated for 2 h.
The absorbance was measured at a wavelength of 450 nm, using a
microplate reader (Model 680, Bio-Rad Laboratories, Hercules, CA,
USA). Each experiment was repeated three times.
Western blot analysis
Cells lysis, protein extraction and quantification
were performed as previously described (12). Equal amounts of protein (20 μg),
from the harvested cells, was loaded onto 10–15% w/v polyacrylamide
gels and separated by SDS-PAGE, followed by transfer to
polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA).
The membranes were blocked with 5% non-fat milk in
phosphate-buffered saline (PBS), at room temperature for 1 h.
Subsequently, the membranes were incubated overnight at 4°C, with a
1:1,000 dilution of either rabbit polyclonal antibody against LC3
or rabbit monoclonal antibody against Beclin 1 (Cell Signaling
Technology, Danvers, MA, USA), or a horseradish peroxidase
(HRP)-conjugated β-actin monoclonal antibody (1:20,000 dilution;
Sigma-Aldrich). The membranes were then incubated with a
HRP-conjugated anti-rabbit immunoglobulin G (1:6,000 dilution;
Sigma-Aldrich) secondary antibody, for 1 h. The membranes were
washed three times with PBS-Tween®, between each
antibody incubation. The protein bands were visualized using an
Enhanced Chemiluminescence Western Blot Analysis system (Pierce
Biotechnology, Inc., Rockford, IL, USA), and quantified by
densitometry using Quantity One Image Analysis Software (Bio-Rad
Laboratories).
siRNA transfection
siRNA sequences specifically targeting human Beclin
1, and non-target control sequences were constructed by Genepharma
Co., Ltd. (Shanghai, China). The sequences used were as follows:
Beclin 1 siRNA sense, 5′-CGGCUCCUAUUCCAUCAAATT-3′, and anti-sense,
5′-UUUGAUGGAAUAGGAGCCGTT-3′; control siRNA sense,
5′-UUCUCCGAACGUGUCACGUTT-3′, and anti-sense,
5′-ACGUGACACGUUCGGAGAATT-3′. A total of 2×105 cells/well
were seeded into 6-well plates, and the following day were
transfected with 100 nM final concentration Beclin 1 siRNA, using
Lipofectamine® 2000 reagent (Invitrogen Life
Technologies, Carlsbad, CA, USA), according to the manufacturer’s
instructions. The cells were collected 48 h following transfection,
for cell viability and apoptosis assays.
Immunofluorescence
The A2780 and A2780cp cells were grown on round
glass coverslips (Fisher Scientific, Waltham, MA, USA) in 35 mm
cell culture dishes. Following a 20 min fixation with pre-chilled
methanol, the coverslips were washed with PBS, permeabilized with
0.2% Triton X-100-PBS for 15 min, and blocked with 2% bovine serum
albumin-PBS for 30 min. The coverlips were then incubated with
rabbit polycolonal p62 and goat polyclonal LC3 primary antibodies
(1:50) at 37°C for 90 min in the dark, followed by three 10 min
washes in PBS. Subsequently, the coverlips were incubated with
goat-anti rabbit IgG-TR and donkey anti-goat IgG-FITC secondary
antibodies, respectively (1:1,000) at 37°C for 1 h in the dark, and
washed three times (10 min/wash) with PBS. All of the antibodies
were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). The
coverslips were mounted onto glass slides, with antifade mounting
medium purchased from Invitrogen (Paisley, UK). The images were
captured using a Zeiss Observer.Z1 microscope (Zeiss, Oberchoken,
Germany) and Slidebook 4.2.0.11 computer software (Intelligent
Imaging Innovations, Inc., Denver, CO, USA).
Transmission electron microscopy
The cells were fixed using 2.5% glutaraldehyde in
0.1 M phosphate buffer for 2 h at 4°C, and then post-fixed using 1%
osmium tetroxide for 3 h. The samples were scraped and pelleted,
dehydrated in a graded series of ethanol baths, infiltrated, and
embedded in Epon™ resin. Ultrathin sections (70 nM) were cut using
a Leica Ultracut Microtome (Leica Microsystems Inc., Buffalo Groce,
Il, USA), stained with uranyl acetate for 3 min, and examined using
a JEOL JEM-1400 transmission electron microscope (JEOL Ltd., Tokyo,
Japan).
Apoptosis analysis
For the assessment of the cellular apoptotic rate,
the fluorescein isothiocyanate Annexin V Apoptosis Detection kit I
(BD Pharmingen, San Diego, CA, USA) was used. Following 48 h of
treatment, the cells were collected and centrifuged at 3,190 × g
for 5 min. The cells were then resuspended in 500 μl binding buffer
and stained with 5 μl Annexin V and 5 μl propidium iodide (PI), for
15 min at room temperature in the dark. The samples were analyzed
by flow cytometric analysis (FC500 MPL, Beckman Coulter, Brea, CA,
USA). A total of 2,000 events were measured and the results are
presented as the percentage (Annexin V positive) of apoptotic
cells.
Statistical analysis
The data are presented as the means ± standard
deviation. A two-tailed student’s t-test was used to compare the
differences between two groups. The analyses were performed using
SPSS version 16.0 (SPSS, Inc., Chicago, IL, USA) software.
P<0.05 was considered to indicate a statistically significant
difference.
Results
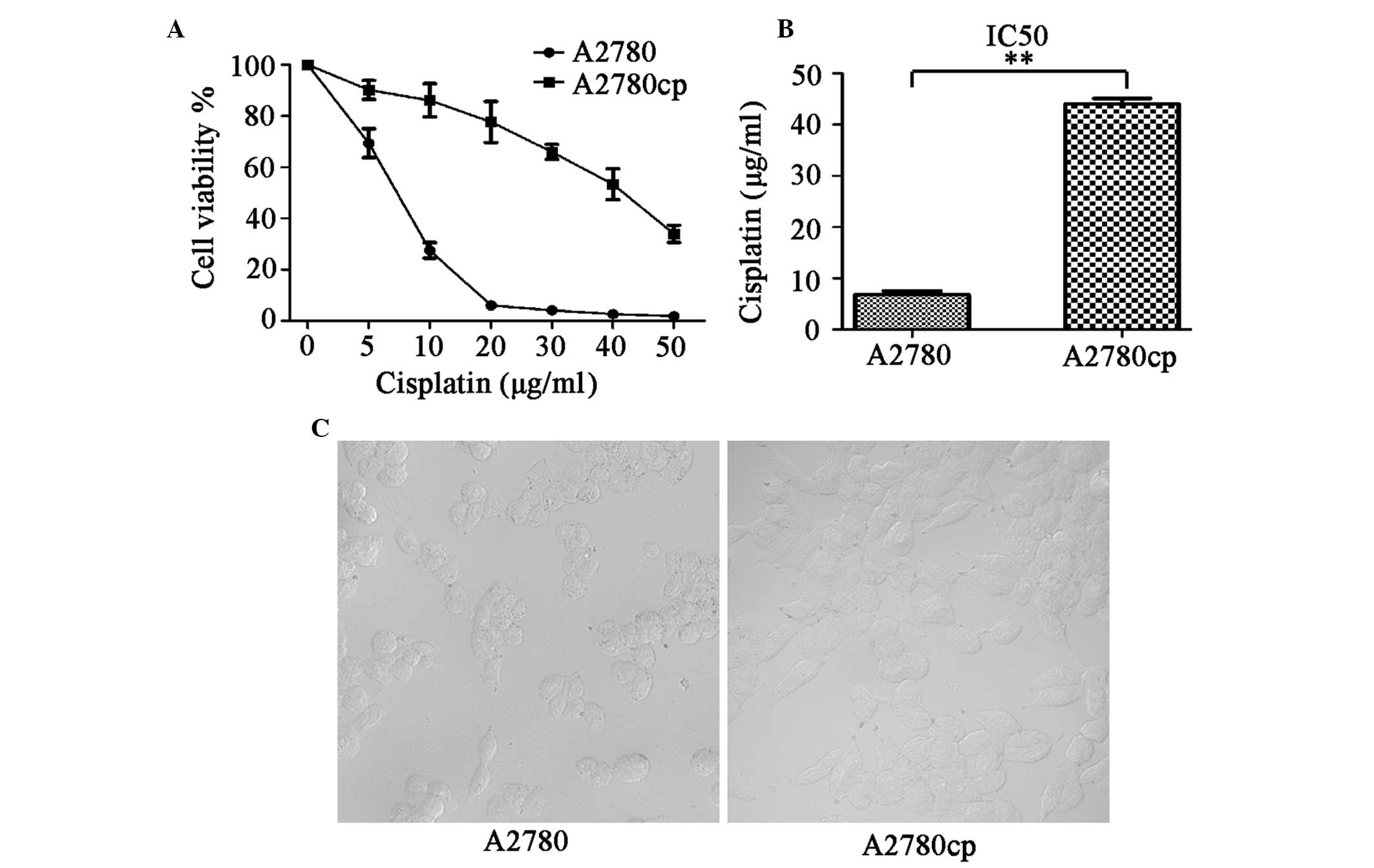
A2780cp cells are resistant to
cisplatin-induced cell death
To verify that the A2780cp cells were resistant to
cisplatin, the parental A2780 and A2780cp cells were treated with
increasing concentrations of cisplatin (5–50 μg/ml) for 24 h, and
the cell viability was measured using a WST-8 assay. The percentage
of surviving cells decreased in a dose-dependent manner in both the
A2780 and A2780cp cells (Fig. 1A).
However, as expected, the A2780cp cells were 6.5× more resistant to
cisplatin, as compared with the A2780 parental cells (P<0.01).
The 24 h half maximal inhibitory concentrations (IC50)
of cisplatin in A2780cp and A2780 cells, were 44.07±1.1 and
6.84±0.66 μg/ml, respectively (Fig.
1B). Using phase-contrast microscopy, the A2780cp cells were
observed as having a regular, round shape, and were markedly larger
as compared with the A2780 cells (Fig.
1C).
Autophagy is involved in cisplatin
resistance in ovarian cancer cells
The endogenous levels of autophagy were compared in
the A2780cp cisplatin-resistant and A2780 sensitive cell lines, by
measuring the protein expression levels of LC3 II and Beclin 1.
A2780cp cells exhibited higher expression levels of LC3 II and
Beclin 1 proteins, as compared with the A2780 cells (Fig. 2A, lanes 1 and 5).
Immunofluorescence staining for LC3 and p62, another
autophagy-related protein, also showed that the cisplatin-resistant
cells exhibited higher levels of autophagy (Fig. 2B). These findings suggest that
increased levels of autophagy may contribute to cisplatin
resistance in ovarian cancer cells.
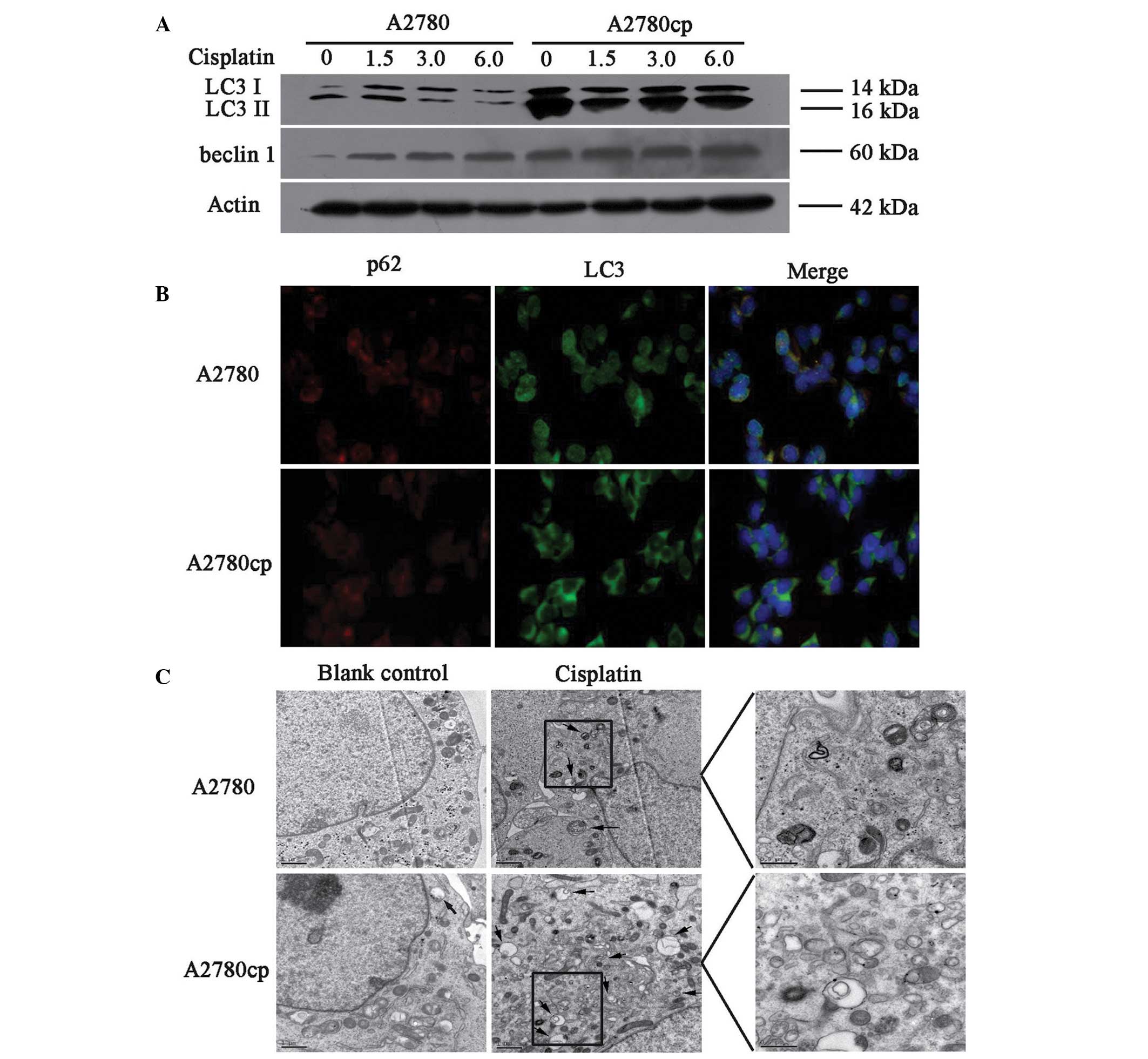 | Figure 2Autophagy induced by cisplatin
treatment in A2780 and A2780cp ovarian cancer cells. (A) The cells
were treated with different concentrations of cisplatin (0, 1.5, 3,
6 μg/ml) for 24 h. The cell lysates were collected for western blot
analysis using antibodies against microtubule-associated protein 1
light chain 3 (LC3) and Beclin 1. (B) Indirect immunofluorescence
of LC3 and p62 was performed in both cell lines, the red signal
represents the p62 levels, and the green signal represents the LC3
levels (magnification, 40x). (C) Representative electron microscopy
images of autophagosomes (magnification, 10,000x). The scale bars
represent 1 μm, and the arrows indicate the autophagosomes. kDa,
kilodaltons. |
The present study also determined whether cisplatin
treatment induced autophagy in both of the cell lines. The cells
were treated with 1.5, 3 and 6 μg/ml cisplatin for 24 h, and then
the protein expression levels of LC3 and Beclin 1 were determined.
As shown in Fig. 2A, cisplatin
induced the protein expression of LC3 II and Beclin 1 in both cell
lines. Notably, the protein expression levels of Beclin 1 in both
cell lines was increased in a dose-dependent manner, whereas LC3 II
did not change (Fig. 2B and C).
Electron microscopic analyses also revealed that treatment with
cisplatin increased the amount of autophagosomes in both cell
lines; however, the number of autophagosomes increased to a greater
extent in the A2780cp cells, as compared with the A2780 cells
(Fig. 2C). These findings suggest
that cisplatin may induce autophagy in ovarian cancer cell lines,
and the induced level of autophagy was higher in A2780cp cells, as
compared with that in A2780 cells.
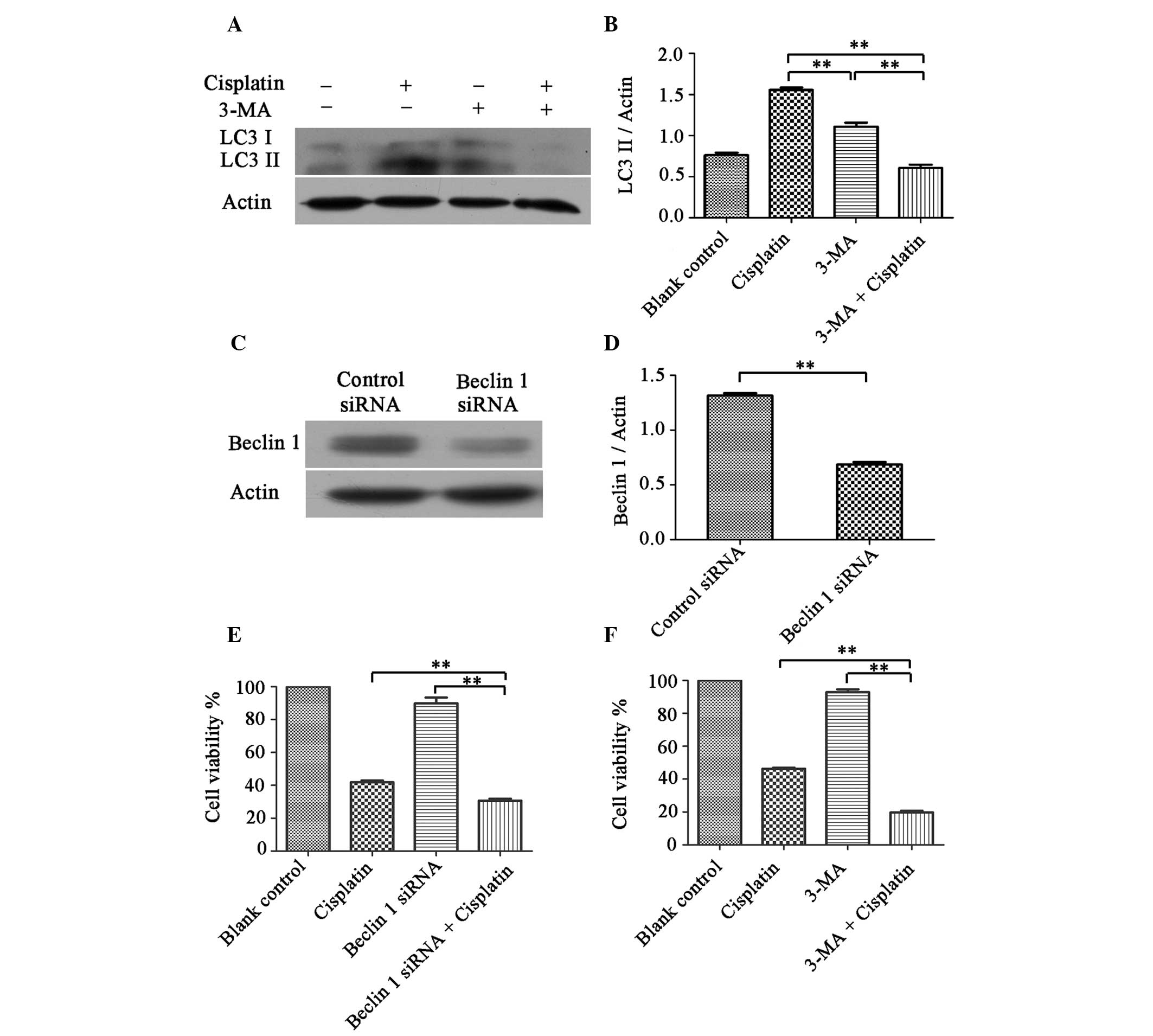
Inhibition of autophagy sensitizes cells
to cisplatin treatment
A2780cp cells were shown to have elevated levels of
autophagy. Therefore, to determine whether they could be
re-sensitized to cisplatin, the cells were treated with an
inhibitor of autophagy, 3-MA, or a Beclin 1 targeting siRNA.
A2780cp cells were pretreated with 1 mmol 3-MA for 1 h, followed by
cisplatin (6 μg/ml) for 24 h. The cell lysates were subjected to
western blot analysis, to determine the protein expression levels
of LC3 I and LC3 II. As shown in Fig.
3A and B, treatment with 3-MA decreased the LC3 II protein
expression levels induced by cisplatin and increased
cisplatin-induced cell death in A2780cp cells (Fig. 3F). As shown in Figure 3C and D, the Beclin 1 siRNA
treatment group had a ~50% reduction in Beclin 1 expression, as
compared with the control siRNA treatment group. Furthermore,
knockdown of Beclin 1 expression sensitized A2780cp cells to
cisplatin, by enhancing cisplatin-induced cell death (Fig. 3E).
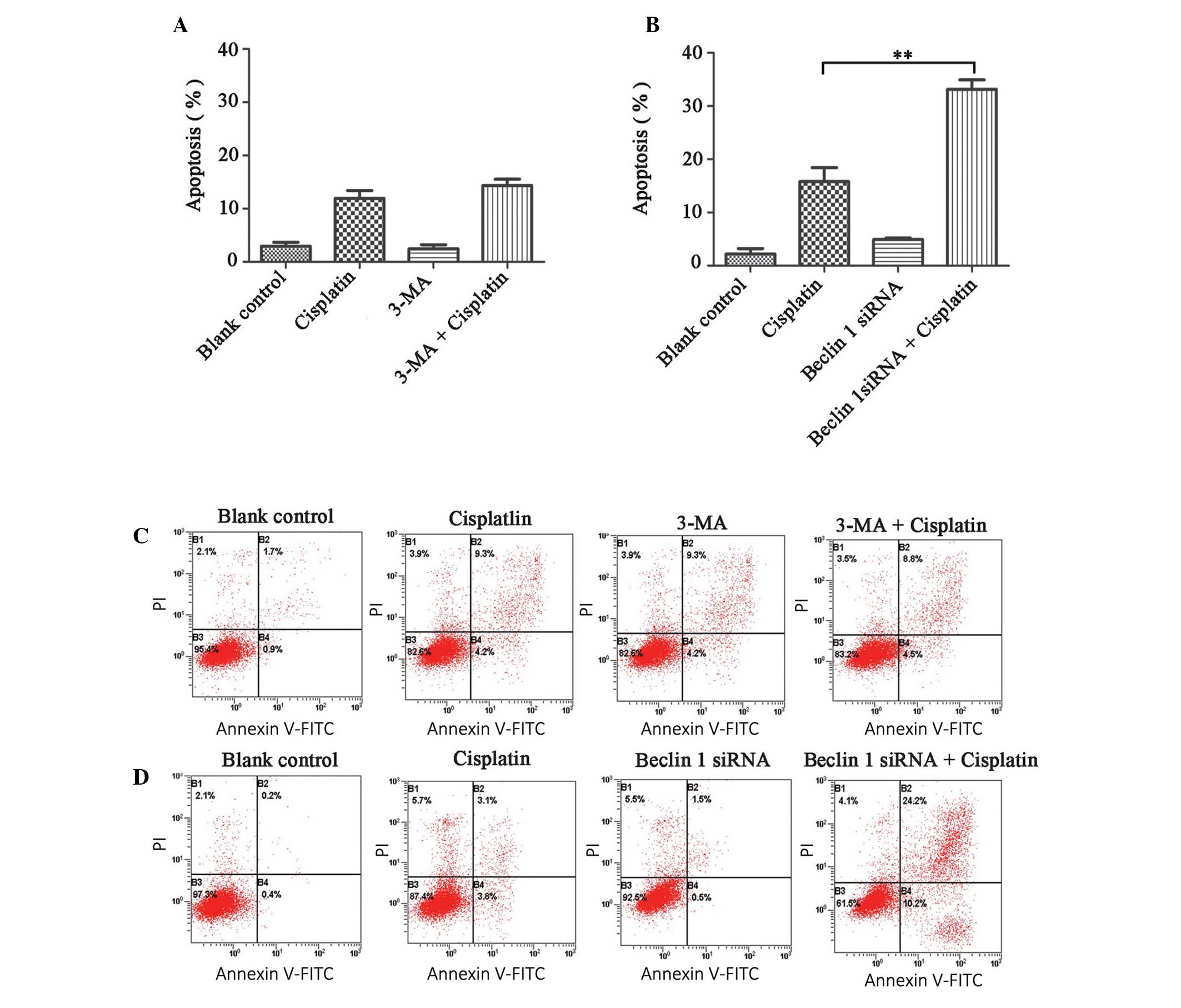
Inhibition of autophagy enhances
cisplatin-induced apoptosis
To determine whether the inhibition of autophagy
influenced cisplatin-induced apoptosis in A2780cp cells, an
apoptosis assay was performed following a co-treatment of
cisplatin, with either 3-MA or Beclin 1 siRNA transfection. The
apoptotic rate (at both the early and advanced stages) of the
control, 3-MA and Beclin 1 siRNA groups were 2.96±0.72, 2.43±0.79
and 4.92±0.28%, respectively (Fig. 4A
and B). Furthermore, the 3-MA plus cisplatin group did not
increase the apoptotic rate of the cells, as compared with the
cisplatin only group (14.35±1.78 vs. 11.91±1.49%, P>0.05;
Fig. 4A and C). However, the
Beclin 1 siRNA plus cisplatin group, had an increased percentage of
apoptotic cells, as compared with the cisplatin only group
(33.14±1.78 vs. 15.79±2.62%, P<0.05, Fig. 4B and D).
Discussion
Cisplatin resistance is a major obstacle in the
successful treatment of cancer, including ovarian cancer. Previous
studies have attempted to elucidate the mechanisms responsible for
cisplatin resistance in cancer. A prominent hypothesis suggests
that resistant cells fail to undergo apoptosis following cisplatin
treatment. Consequently, numerous studies have attempted to use
anti-apoptotic inhibitors, to sensitize resistant cancer cells and
tumors to chemotherapeutics. However, these studies have
demonstrated that targeting apoptosis does not optimally inhibit
chemoresistance (13,14). Therefore, alternative cell-death
pathways, such as autophagy, have become an important area of
research; however, the role of autophagy in cancer chemoresistance
remains unclear. Previously, a study using SKOV3 ovarian cancer
cells, revealed that cisplatin-resistant ovarian cancer cells
expressed high levels of autophagy. However, this study focused on
the ubiquitin protein p62, which was shown to regulate autophagy
degradation, prevent endoplasmic reticulum stress-induced apoptosis
and lead to cisplatin resistance in human ovarian cancer cells, but
provided limited information on the role of autophagy in cisplatin
resistance (15).
To further explore the role of autophagy in
cisplatin resistance, the present study used A2780 and A2780cp
cells as a model for in vitro analysis. A cell viability
assay confirmed that A2780 and A2780cp cells provide an ideal pair
of cell lines to use for these studies, since A2780cp cells were
6.5× more resistant to cisplatin, as compared with the parental
cell line. The level of autophagy was evaluated in both the ovarian
cancer cell lines. Autophagy is regulated through a family of ATG
genes. Beclin 1, a mammalian autophagy gene, is generally combined
with class III phosphoinositide 3-kinase, as a complex which has
been shown to be necessary for the initiation of autophagy
(16). During the formation of an
autophagosome, LC3 is cleaved to produce its active form: LC3 I,
which conjugates with phosphatidylethanolamine to form LC3 II,
which is localized to the autophagosomal membrane (17). Therefore, LC3 II may be examined as
an indicator of autophagy activity. P62 is a polyubiquitin-binding
protein, which binds directly to LC3 and is degraded by autophagy
activation (18,19). In the present study, following the
treatment of the cells with different concentrations of cisplatin
for 24 h, both autophagy markers, LC3 and Beclin 1 were upregulated
in A2780cp cells, as compared with the A2780 cells, as determined
by western blot analysis. Furthermore, untreated A2780cp cells
expressed greater amounts of LC3 and a lower amount of p62, as
determined by immunofluorescence, as compared with the A2780 cells.
These results suggested that autophagy was more active in
cisplatin-resistant cells. The previous SKOV3 ovarian cancer cell
study examined the accumulation of LC3 by western blot analysis, to
distinguish autophagy levels in the cells (15). To confirm that chemoresistant
ovarian cancer cells expressed higher levels of autophagy, LC3 and
Beclin 1 protein expression levels were evaluated by western
blotting, alongside the amount of p62 and LC3 through indirect
immunofluorescence, and the number of autophagosomes by
transmission electron microscopy. The levels of autophagy increased
in response to cisplatin, in a dose dependent manner, in the
A2780cp resistant cells. These results suggested that there is a
protective role of autophagy in cisplatin resistance.
To explore whether inhibiting autophagy may
sensitize resistant cells to cisplatin treatment, the effects of an
inhibitor of autophagy or Beclin 1 siRNA were examined on cell
death, in A2780cp cells. 3-MA is a specific inhibitor of the
autophagic pathway, which functions by inhibiting the class III
phosphatidylinositol 3-kinases and blocking the formation of
autophagosomes at the sequestration step (20,21).
Previously, in EC9706 esophageal squamous carcinoma cells, 3-MA was
shown to contribute to the upregulation of cisplatin-induced cell
death (22). In the present study,
low doses of 3-MA (1 mmol) suppressed LC3 II protein formation and
sensitized A2780cp cells to cisplatin treatment. Beclin 1 has a
critical, regulatory role in autophagy, and downregulation of
Beclin 1 has previously been shown to sensitize Hela human cervical
cancer cells and HepS mouse liver cancer cells to cisplatin
(23). In the present study,
knockdown of Beclin 1 expression, using siRNA, significantly
inhibited autophagy and sensitized A2780cp cells to cisplatin
treatment. Furthermore, the apoptotic rate of the cells was
analyzed, to explore the potential mechanisms of autophagy
inhibition on cisplatin sensitization. Previously, in HT29 human
colorectal cancer cells, 3-MA treatment enhanced 5-FU-induced
apoptosis (24). In the present
study, however, the apoptotic rates were not markedly altered
following a co-treatment of cisplatin with 3-MA. Similar effects
were observed in esophageal cancer cells, in which disruption of
lysosomal activity with the pharmacological inhibitors bafilomycin
A1 or chloroquine did not improve the chemotherapeutic effects
(9). A possible explanation is
that pharmacological inhibitors exert transient effects, which may
result in the activation of another potential cell-death mechanism,
induced by cisplatin. However, knockdown of Beclin 1 expression,
with siRNA, significantly inhibited autophagy and increased
cisplatin-induced apoptosis. This finding is consistent with a
previous study, which indicated that the cleavage of Beclin 1
reduced autophagy and promoted apoptosis in HeLa cells (25). These findings suggest that Beclin 1
may be a potential target for autophagy inhibition, in order to
sensitize cancer cells to chemotherapy.
In conclusion, higher levels of autophagy were
observed in cisplatin-resistant ovarian cancer cells, whereas
knockdown of Beclin 1 expression restored cisplatin-sensitivity to
these cells, which is attributed to autophagy inhibition.
Therefore, targeting autophagy may be a potential therapeutic
strategy in ovarian cancer treatment.
Acknowledgements
The present study was funded by the National Natural
Science Foundation of China (No. 81302261) and the Natural Science
Foundation of Science and Technology Commission of Shanghai
Municipality (No. 14411965700).
References
|
1
|
Kim A, Ueda Y, Naka T and Enomoto T:
Therapeutic strategies in epithelial ovarian cancer. J Exp Clin
Cancer Res. 31:142012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siddik ZH: Cisplatin: mode of cytotoxic
action and molecular basis of resistance. Oncogene. 22:7265–7279.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Galluzzi L, Senovilla L, Vitale I, et al:
Molecular mechanisms of cisplatin resistance. Oncogene.
31:1869–1883. 2012. View Article : Google Scholar
|
|
4
|
Carew JS, Nawrocki ST and Cleveland JL:
Modulating autophagy for therapeutic benefit. Autophagy. 3:464–467.
2007. View Article : Google Scholar
|
|
5
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar
|
|
6
|
Eskelinen EL and Saftig P: Autophagy: a
lysosomal degradation pathway with a central role in health and
disease. Biochim Biophys Acta. 1793:664–673. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guo XL, Li D, Hu F, et al: Targeting
autophagy potentiates chemotherapy-induced apoptosis and
proliferation inhibition in hepatocarcinoma cells. Cancer Lett.
320:171–179. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
O’Donovan TR, O’Sullivan GC and McKenna
SL: Induction of autophagy by drug-resistant esophageal cancer
cells promotes their survival and recovery following treatment with
chemotherapeutics. Autophagy. 7:509–524. 2011.PubMed/NCBI
|
|
10
|
Wu MY, Fu J, Xu J, O’Malley BW and Wu RC:
Steroid receptor coactivator 3 regulates autophagy in breast cancer
cells through macrophage migration inhibitory factor. Cell Res.
22:1003–1021. 2012. View Article : Google Scholar
|
|
11
|
Sirichanchuen B, Pengsuparp T and
Chanvorachote P: Long-term cisplatin exposure impairs autophagy and
causes cisplatin resistance in human lung cancer cells. Mol Cell
Biochem. 364:11–18. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bao LJ, Jaramillo MC, Zhang ZB, et al:
Nrf2 induces cisplatin resistance through activation of autophagy
in ovarian carcinoma. Int J Clin Exp Pathol. 7:1502–1513.
2014.PubMed/NCBI
|
|
13
|
Wang CY, Cusack JC Jr, Liu R and Baldwin
AS Jr: Control of inducible chemoresistance: enhanced anti-tumor
therapy through increased apoptosis by inhibition of NF-kappaB. Nat
Med. 5:412–417. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gallego MA, Joseph B, Hemström TH, et al:
Apoptosis-inducing factor determines the chemoresistance of
non-small-cell lung carcinomas. Oncogene. 23:6282–6291. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu H, Su J, Xu Y, et al: p62/SQSTM1
involved in cisplatin resistance in human ovarian cancer cells by
clearing ubiquitinated proteins. Eur J Cancer. 47:1585–1594. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kihara A, Kabeya Y, Ohsumi Y and Yoshimori
T: Beclin-phosphatidylinositol 3-kinase complex functions at the
trans-Golgi network. EMBO Rep. 2:330–335. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kabeya Y, Mizushima N, Ueno T, et al: LC3,
a mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pankiv S, Clausen TH, Lamark T, Brech A,
Bruun JA, Outzen H, Øvervatn A, Bjørkøy G and Johansen T:
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mathew R, Karp CM, Beaudoin B, et al:
Autophagy suppresses tumorigenesis through elimination of p62.
Cell. 137:1062–1075. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Seglen PO and Gordon PB: 3-Methyladenine:
specific inhibitor of autophagic/lysosomal protein degradation in
isolated rat hepatocytes. Proc Natl Acad Sci USA. 79:1889–1892.
1982. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stroikin Y, Dalen H, Lööf S and Terman A:
Inhibition of autophagy with 3-methyladenine results in impaired
turnover of lysosomes and accumulation of lipofuscin-like material.
Eur J Cell Biol. 83:583–590. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu D, Yang Y, Liu Q and Wang J:
Inhibition of autophagy by 3-MA potentiates cisplatin-induced
apoptosis in esophageal squamous cell carcinoma cells. Med Oncol.
28:105–111. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zou Z, Wu L, Ding H, et al: MicroRNA-30a
sensitizes tumor cells to cis-platinum via suppressing Beclin
1-mediated autophagy. J Biol Chem. 287:4148–4156. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li J, Hou N, Faried A, Tsutsumi S,
Takeuchi T and Kuwano H: Inhibition of autophagy by 3-MA enhances
the effect of 5-FU-induced apoptosis in colon cancer cells. Ann
Surg Oncol. 16:761–771. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhu Y, Zhao L, Liu L, et al: Beclin 1
cleavage by caspase-3 inactivates autophagy and promotes apoptosis.
Protein Cell. 1:468–477. 2010. View Article : Google Scholar : PubMed/NCBI
|