Introduction
Cri du chat syndrome (CdCS), which stems from a
partial deletion of the short arm of chromosome 5 (5p-), is a rare
genetic disorder occuring in ~1/15,000 to 1/50,000 live births
(1), though it is more commonly
identified in patients with mental retardation (~1/350) (1). As suggested by the name, CdCS is
typically characterized by the occurrence of a high-pitched,
cat-like cry, though this is lacking in certain cases (2). Therefore, the phenotype of CdCS may
be divided into two categories, one for the typical cat-like cry
and another for the other clinical traits. In principle, these two
distinct phenotypes imply the existence of more than one critical
region in 5p (3); differences in
the sizes of deletions spanning these regions are likely
responsible for some of the variability of CdCS.
While ~90% of cases are attributed to a sporadic or
de novo deletion with random occurrence, a few arise from
the malsegregation of a balanced translocation in the parental
karyotype (4). Under this
condition, the 5p monosomy tends to be accompanied by a partial
trisomy of the genome. A priori, these patients may exhibit
more severe phenotypes than those with only monosomy of 5p. In the
case presented here, a trisomic portion was identified at
chromosome 18.
Trisomy 18, or Edwards syndrome, is the second most
common autosomal trisomy syndrome following trisomy 21 (Downs
syndrome), and it results from full, mosaic, or partial trisomy of
18q. This syndrome has a very high mortality rate due to heart
abnormalities, kidney malformations and other internal organ
disorders. While full trisomy of 18q is the most prevalent form
(5–8), partial trisomy of the terminal region
of 18q is a very rare form that may present nonspecific
abnormalities, including intrauterine growth restriction,
microcephaly, a prominent forehead, hypertelorism, a short neck,
mental retardation, seizure, laryngomalacia, atrial stenosis and
club foot (9–12).
The deletion of 5p and partial 18q trisomy are rare
occurances. The purpose of current study was to report an example
of this unique combination in a female patient with an unbalanced
translocation, giving rise to 5p deletion and 18q duplication, in
addition to determining whether this case may assist in confirming
critical regions of 5p that have previously been reported to cause
typical CdCS, and whether partial trisomy of 18q influences the
clinical characteristics, even though the CdCS phenotype prevails.
To address these issues, the karyotype and phenotype of the patient
and her parents were assessed using G-banding techniques,
chromosome microarray analysis (CMA), and fluorescence in
situ hybridization (FISH). The regions involved in or the
extent of the 5p deletion and 18q duplication are good candidates
for mapping the phenotypic disruptions associated with CdCS and
Edwards syndrome. Therefore, determining which region has the
genetic disorder in this case is of great clinical and etiological
value.
Materials and methods
Patient data
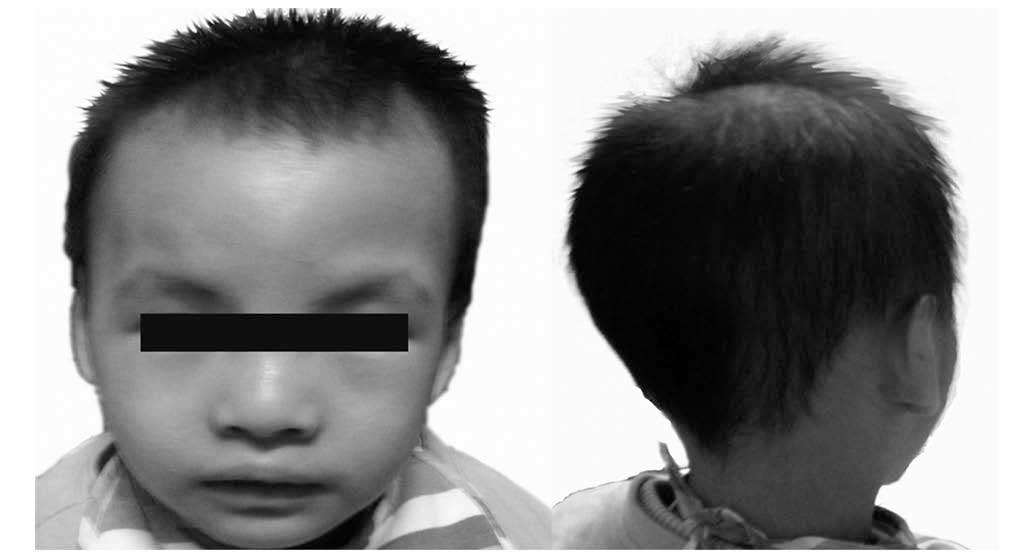
The two-year-old female patient was born at 39
weeks. She presented with laryngomalacia, atrial septal defect, a
downturned mouth, transverse flexion creases and hypotonia, all of
which are typical phenotypes of CdCS. In addition, the patient
displayed psychomotor retardation, developmental delay and a long
face, phenotypes which become evident with developing CdCS,
however, she also had almond-shaped eyes and a bulbous nose
(Fig. 1). Parental karyotyping
detected an apparent balanced translocation (46, XY,
t(5;18)(p13;q22)) in the father and no evident chromosomal
abnormalities in the mother. Samples and pictures from the patient
and her family were obtained following informed consent. This study
was approved by the ethics committee of The First Affiliated
Hospital of Sun Yat-sen University, Guangzhou, China.
Conventional cytogenetic analysis
A 5 ml blood sample was collected from the patient
and each of her parents. Lymphocytes were cultured from each sample
and used for cytogenetic analysis according to the standard blood
cytogenetic protocol (13).
Further routine cytogenetic analysis was performed using G-banding
at 550 band resolution (trypsin: Amresco, Solon, OH, USA; giemsa
stain: Sigma-Aldrich, St. Louis, MO, USA).
CMA
CMA was performed using the Affymetrix cyto HD Array
(Affymetrix, Santa Clara, CA, USA). DNA was amplified, labeled and
hybridized to the CytoScan HD array platform according to the
manufacturer’s instructions. The array is specifically designed for
cytogenetics research and offers >2 million markers across the
genome, including single nucleotide polymorphism probes and probes
to detect copy number variations (Cyto-arrays). CEL files obtained
by scanning CytoScan arrays were analyzed with the Chromosome
Analysis Suite software (Affymetrix), employing genome annotation
data (version GRCH37, hg19). Only data achieving the manufacturer’s
quality cut-off levels were included in further analyses.
Primarily, gains and losses that affected a minimum of 50 markers
within a 100 kbp length were accepted.
FISH
The FISH test for confirming the der(5)t(5:18) of the derivative chromosome was
conducted using the Vysis Cri-du-Chat region probe (Vysis, Downers
Groove, IL, USA) and a combination of the CEP18 probes and 18q
subtelomere-specific probes (Vysis) as described in the
manufacturer’s protocol, using the standard FISH protocol (14).
Results
CMA
The array detected two genomic anomalies in the
patient’s genome: a 32 Mb deletion at 5p15.33-p13.3 (chr 5: 1–32,
137, 848) (Fig. 2A) and a 6.6 Mb
duplication at 18q22.3q23 (chr 18: 71, 368, 578–78, 014, 123)
(Fig. 2B). The deletion
encompasses the complete terminal region of chromosome 5 covering
band 5p15.2 and band 5p15.3, which are well established CdCS
critical regions. In contrast, a review of the literature shows the
duplication, which resides in the terminal region of 18q, is not
known to be associated with abnormal genotypes, appearing in
phenotypically normal males (15).
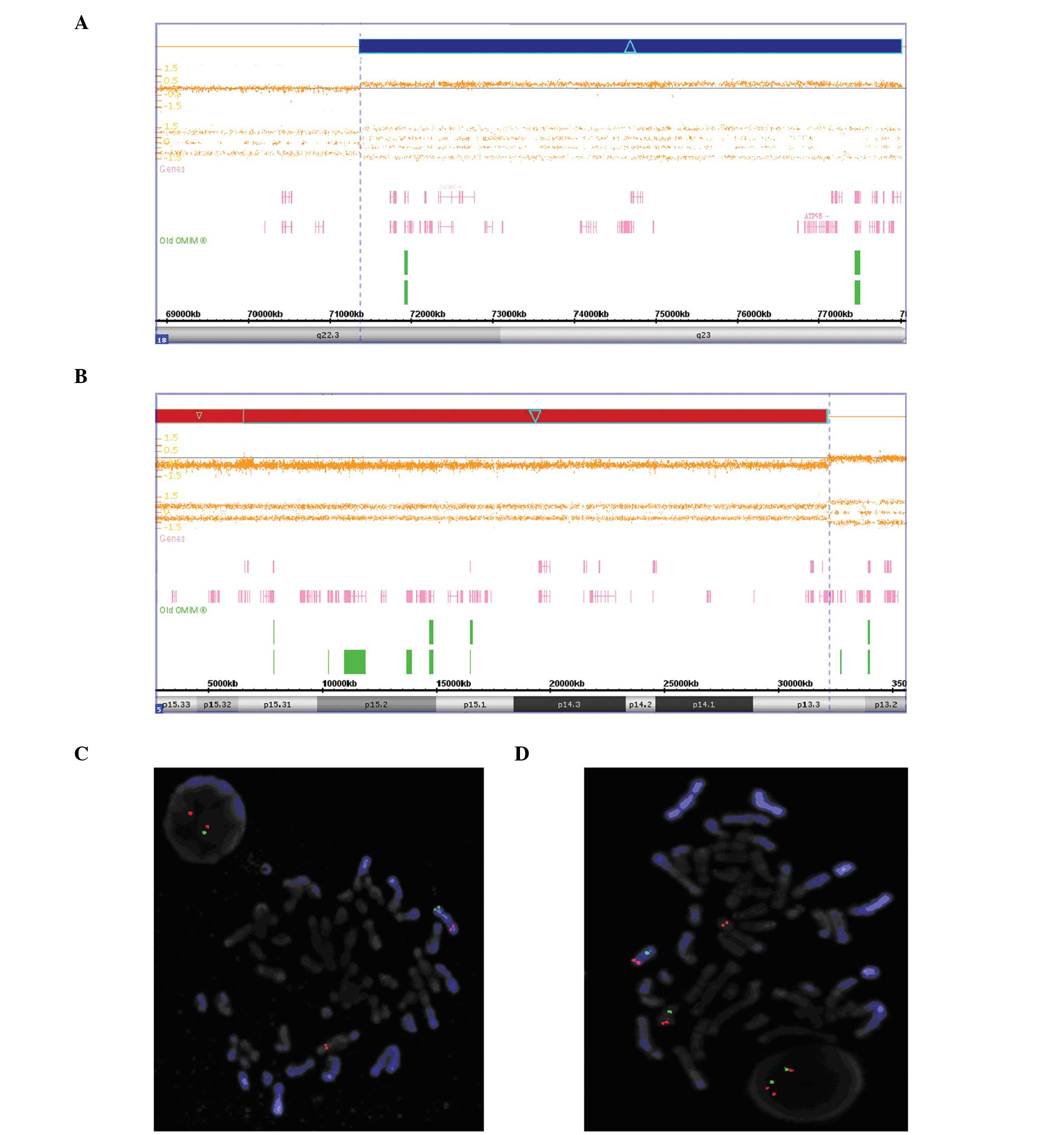 | Figure 2A deletion of 5p and a duplication of
18q analyzed using Chromosome Analysis Suite software. The array
detected two genomic anomalies in the patient’s genome, (A) a 32 Mb
deletion at 5p15.33-p13.3 (chr 5: 1–32, 137,848) and (B) a 6.6 Mb
duplication at 18q22.3q23 (chr 18: 71, 368,578–78,014,123). FISH
results confirmed the deletion of Chr5 and the duplication of
Chr18. Using the Vysis Cri-du-chat region probe, FISH analysis of
metaphase cells taken from the proband showed one copy of
chromosome 5 containing one red and one green signal, whereas its
counterpart only contained one red signal, suggesting it is the
derivative chromosome 5 or der (5). (C) Interphase cells showed two red
signals and one green signal. Further FISH analysis for chromosome
18 confirmed balanced translocation of karyotype, with metaphase
cells showing a red signal (18q telomeric probe)on one copy of
chromosome 18 and a red signal on the other copy of chromosome 18
compared with both one green(CEP18 alpha satellite probe)and one
red signal on the derivative chromosome 5 or der (5). (D) Interphase cells showed two green
signals and three red signals. The labeled chromosomes 5, der
(5), and 18 in the metaphase FISH
pictures are identified by DAPI-banding using an ASI FishVision
workstation (Applied Imaging). Magnification, ×1,000. FISH,
fluorescence in situ hybridization. |
FISH analysis
FISH analysis of the proband and her parents
confirmed the rearrangements and revealed that the partial monosomy
of 5p and the partial trisomy of 18q were the result of a
reciprocal unbalanced malsegregation derived from her father
(Fig. 2C and D).
Comparative clinical features
The clinical phenotypes of the current patient, who
carries these two rare genomic imbalances, were compared with the
phenotypes of patients carrying only one of these genomic
imbalances. The results are summarized in Tables I and II. While mental retardation or
developmental delay was consistently observed in all patients with
partial 5p monosomy, the present patient also presented a number of
physical features that are not frequently observed in other
patients, including hypotonia and a bulbous nose. Microcephaly was
observed in certain patients and is a common phenotype of CdCS;
however, it was not observed in the present case. In addition, the
current study determined that the patient presented features that
have not previously been described in other patients, such as
transverse flexion creases, a downturned mouth, a long face and
almond-shaped eyes. The more severe features observed in this
patient may stem from the deletion at 5p15.33-p13.3, which is
larger than deletions found in other patients. They may also be
caused by the presence of the 18q duplication. However, patients
with partial chromosome 18 trisomy alone did not exhibit the
abnormalities observed in the current case, although mental
retardation or developmental delay was a common feature in those
patients. This suggests that in the present case, the 18q
duplication may have a weaker effect on the phenotype than the
larger 5p deletion.
 | Table IPhenotypic comparison of patients with
pure partial chromosome 5 monosomy. |
Table I
Phenotypic comparison of patients with
pure partial chromosome 5 monosomy.
| Decipher ID | | |
|---|
|
| | |
|---|
| Category | 258252 | 276535 | 272684 | 270177 | 268106 | Typical Cri du Chat
Syndrome | Present case |
|---|
| Size (Mb) | 11.10 Mb | 10.88 Mb | 22.09 MB | 8.64 Mb | 14.25 Mb | 12.52 Mb | 32 Mb |
| Region | 95302-11199684 | 95243-10972789 | 151737-22246071 | 22179-8659713 | 3209639-17455995 | 10001-12533304 | 1-32137848 |
| Inheritance | Unknown | Unknown | Unknown | de novo | de novo | − | Parental |
| Weak high-pitched
voice | − | − | − | − | − | + | + |
| Mental
impairment | + | + | + | + | + | + | + |
| Transverse flexion
creases | − | − | − | − | − | − | + |
| Downturned mouth | − | − | − | − | − | − | + |
| Hypotonia | − | + | − | − | − | − | + |
| Long face | − | − | − | − | − | − | + |
| Almond-shaped
eyes | − | − | − | − | − | − | + |
| Bulbous nose | − | − | − | − | + | − | + |
| Microcephaly | − | + | − | − | + | + | − |
| Table IIPhenotypic comparison of patients with
pure partial chromosome 18 trisomy. |
Table II
Phenotypic comparison of patients with
pure partial chromosome 18 trisomy.
| Decipher ID | |
|---|
|
| |
|---|
| Category | 3793 | 255714 | 259905 | 266270 | 268116a | Present case |
|---|
| Size (Mb) | 14.83 Mb | 18.53 Mb | 5.93 Mb | 77.92 Mb | 7.37 Mb | 6.6 Mb |
| Region | 178428-15008636 | 14116-18540053 | 362803-6288431 | 83701-78001525 | 363264-7730691 |
71368578-78014123 |
| Inheritance | de novo | Unknown | de novo | Unknown | Unknown | Parental balanced
translocation |
| Weak high-pitched
voice | − | − | − | − | − | + |
| Mental
impairment | + | + | + | − | − | + |
| Transverse flexion
creases | − | − | − | + | | |
| Downturned
mouth | − | − | − | − | − | + |
| Hypotonia | + | − | − | + | − | + |
| Long face | − | − | − | − | − | + |
| Almond-shaped
eyes | − | − | − | − | − | + |
| Bulbous nose | − | − | − | − | − | + |
| Microcephaly | − | − | − | − | − | − |
Discussion
In the patient, a large (32 Mb) terminal deletion of
5p was detected, covering all the CdCS critical regions that had
been reported to date (p15.3, p15.2, p15.1, p14 and p13). The
deletion of p15.3 is the direct cause of typical CdCS (16), while deletion of p15.2 is another
cause of CdCS that does not cause the characteristic catlike cry
(2). The loss of p15.1, p14 and
p13 has been reported in individual families with interstitial
deletions (2,4), varying to different extents in
different individuals. In the current case, the clinical phenotypes
of 5p aberration conformed for the most part to the features of
CdCS, confirming the critical regions reported in prior
studies.
The region of chromosome 5p13.3 that was deleted in
this patient includes 90 well-characterized genes, in addition to a
number of predicted genes postulated to reside within this region.
These genes are commonly deleted in patients with 5p-deletion
syndrome (17), and are associated
with the common clinical features, including the characteristic
facial features. The clinical abnormalities found by physical
examination of the patient in the present study are primarily the
result of the deletion of these critical 5p regions.
Accompanying the terminal deletion of 5p, a partial
duplication of 18q was detected in this case. In a previous study,
patients with partial trisomy of chromosome 18 displayed severe
Edwards syndrome (18), though
complete duplication of chromosome 18 is typically required for the
pathogenesis of Edwards syndrome. Several authors have attempted to
identify the critical regions responsible for phenotypes in full
trisomy 18 by comparing the clinical features of patients with
various chromosome 18q duplications. A number of candidate critical
regions for Edwards syndrome have been proposed on the basis of a
few individual reports, however no consensus has been reached
regarding the location (11,19–22).
The proposed critical regions associated with dysmorphic features
include 18q11.2, 18q21.1q21.2, and 18q22.3qter; the 18q duplication
(18q22.3q23) reported in the current study partially overlapped
with one of these critical regions. Nevertheless, a phenotypically
normal male patient was reported to have a large duplication of the
terminal 17.4 Mb of chromosome 18, including 18q22.3q23 (15), which contradicts its proposed role
as a critical region. Furthermore, certain authors have argued
against the existence of a critical region due to the interaction
of cis-acting genes from several parts of chromosome 18, which are
necessary to produce the full trisomy 18 phenotype (18). Literature searches for direct
evidence of an effect of 18q22.3q23 duplication suggests no
associations, although certain dysmorphic phenotypes seemingly
linked to this region indicate an association.
While these results suggest a prima facie
conclusion that the region from 18q22.3 to q23 has no effect on
dysmorphic phenotypes, it is still possible that the clinical
features associated with duplication of this region are easily
masked by more severe phenotypes from other chromosomal
aberrations. In contrast, a recent study using high-resolution
array-CGH in 29 patients with 18q deletions concluded that the 4.3
Mb region located within 18q22.3q23 is a critical region for the
‘typical’ 18q-phenotype (23). It
is possible that the critical region defined in cases of deletion
is also partly responsible for the phenotypes classically described
for duplication of 18q. A previous study indicated the existence of
concentration-sensitive genes within 18q22.3q23; to date, 29
well-annotated genes have been found in this region, including the
microsomal cytochrome b5 gene (CYB5A; 613218) and the gene
for phosphatase specific for the C-terminal domain (CTD) of
RNA polymerase II subunit A (CTDP1, *604927). The
CTDP1 gene product is involved in the initiation of gene
expression (24) and may have a
role in linking transcription elongation with splicing (25). It remains difficult to predict the
effect of an increased expression of these genes. A similar patient
to that of the current study was identified in the database
DECIPHER. This patient (256304) had a loss of 36 Mb at chr 5: 130,
931–36, 780, 974, and a gain of 37 Mb at chr 8: 40, 832, 757–77,
966, 288, arising from a balanced parental rearrangement, and
presented with atrioventricular canal defect (https://decipher.sanger.ac.uk/patient/256304). It is
notable that this larger duplication, which includes the segment
duplicated in the present patient, coincides with less-severe
abnormalities, although it is possible that the patient’s
phenotypes were not fully detailed. It is unclear what effect the
18q duplication has in the present patient’s phenotype. We hesitate
to conclude that there is no effect from the 18q22.3-q23
duplication; however, the current study has demonstrated that the
net effect is weak. This may serve as a guide for identifying the
critical regions in Edwards syndrome and for analyzing the
functional areas on chromosome 18.
In the current study, the patient showed a variety
of genetic manifestations that were markedly different from those
typical of CdCS. We report the first clinicopathological
characteristics of a patient with a combined chromosomal disorder
of 5p partial monosomy and 18q partial trisomy. The phenotypes of
5p monosomy displayed in this patient agree with previous reports,
and the 18q partial trisomy may have a weak effect, which is
considerably dwarfed by the severity of CdCS. The correlation
between those unique features determined by the karyotype
identified with CMA and validated by FISH will shed light on the
functional mapping of chromosome 18.
Acknowledgements
The authors would like to thank the patient and her
parents for their cooperation in this study. In addition, thanks go
to Chen Junhong and Huang Ailan for their assistance with patient
recruitment, and Fan Qun for her helpful comments and guidance.
References
|
1
|
Niebuhr E: The Cri du chat syndrome:
epidemiology, cytogenetics, and clinical features. Hum Genet.
44:227–275. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gersh M, Goodart SA, Pasztor LM, et al:
Evidence for a distinct region causing a cat-like cry in patients
with 5p deletions. Am J Hum Genet. 56:1404–1410. 1995.PubMed/NCBI
|
|
3
|
Church DM, Yang J, Bocian M, Shiang R and
Wasmuth JJ: A high-resolution physical and transcript map of the
Cri du chat region of human chromosome 5p. Genome Res. 7:787–801.
1997.PubMed/NCBI
|
|
4
|
Mainardi PC, Perfumo C, Cali A, et al:
Clinical and molecular characterisation of 80 patients with 5p
deletion: genotype-phenotype correlation. J Med Genet. 38:151–158.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Embleton ND, Wyllie JP, Wright MJ, Burn J
and Hunter S: Natural history of trisomy 18. Arch Dis Child Fetal
Neonatal Ed. 75:F38–F41. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Carter PE, Pearn JH, Bell J, Martin N and
Anderson NG: Survival in trisomy 18. Life tables for use in genetic
counselling and clinical paediatrics. Clin Genet. 27:59–61. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Young ID, Cook JP and Mehta L: Changing
demography of trisomy 18. Arch Dis Child. 61:1035–1036. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Goldstein H and Nielsen KG: Rates and
survival of individuals with trisomy 13 and 18. Data from a 10-year
period in Denmark. Clin Genet. 34:366–372. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen CP, Chern SR, Wang TH, et al:
Prenatal diagnosis and molecular cytogenetic analysis of partial
monosomy 10q (10q25.3 - >qter) and partial trisomy 18q (18q23 -
>qter) in a fetus associated with cystic hygroma and ambiguous
genitalia. Prenat Diagn. 25:492–496. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
De Muelenaere A, Fryns JP and Van den
Berghe H: Familial partial distal 18q (18q22–18q23) trisomy. Ann
Genet. 24:184–186. 1981.
|
|
11
|
Turleau C and de Grouchy J: Trisomy 18qter
and trisomy mapping of chromosome 18. Clin Genet. 12:361–371. 1977.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schinzel A: Catalogue of Unbalanced
Chromosome Aberrations in Man. 2nd edition. Walter de Gruyter;
Berlin, Germany: pp. 754–780. 2001
|
|
13
|
Varma RS and Babu A: Human chromosomes :
principles and techniques. McGraw-Hill; New York, NY: 1995
|
|
14
|
Garimberti E and Tosi S: Fluorescence in
situ hybridization (FISH), basic principles and methodology.
Methods Mol Biol. 659:3–20. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Quiroga R, Monfort S, Oltra S, et al:
Partial duplication of 18q including a distal critical region for
Edwards Syndrome in a patient with normal phenotype and
oligoasthenospermia: case report. Cytogenet Genome Res. 133:78–83.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Overhauser J, Huang X, Gersh M, et al:
Molecular and phenotypic mapping of the short arm of chromosome 5:
sublocalization of the critical region for the cri-du-chat
syndrome. Hum Mol Genet. 3:247–252. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cerruti Mainardi P: Cri du Chat syndrome.
Orphanet J Rare Dis. 1:332006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mewar R, Kline AD, Harrison W, et al:
Clinical and molecular evaluation of four patients with partial
duplications of the long arm of chromosome 18. Am J Hum Genet.
53:1269–1278. 1993.PubMed/NCBI
|
|
19
|
Fryns JP, Detavernier F, van Fleteren A
and van den Berghe H: Partial trisomy 18q in a newborn with typical
18 trisomy phenotype. Hum Genet. 44:201–205. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Matsuoka R, Matsuyama S, Yamamoto Y,
Kuroki Y and Matsui I: Trisomy 18q. A case report and review of
karyotype-phenotype correlations. Hum Genet. 57:78–82.
1981.PubMed/NCBI
|
|
21
|
Mücke J, Trautmann U, Sandig KR and Theile
H: The crucial band for phenotype of trisomy 18. Hum Genet.
60:2051982. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Boghosian-Sell L, Mewar R, Harrison W, et
al: Molecular mapping of the Edwards syndrome phenotype to two
noncontiguous regions on chromosome 18. Am J Hum Genet. 55:476–483.
1994.PubMed/NCBI
|
|
23
|
Feenstra I, Vissers LE, Orsel M, et al:
Genotype-phenotype mapping of chromosome 18q deletions by
high-resolution array CGH: an update of the phenotypic map. Am J
Med Genet A. 143A:1858–1867. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Archambault J, Pan G, Dahmus GK, et al:
FCP1, the RAP74-interacting subunit of a human protein phosphatase
that dephosphorylates the carboxyl-terminal domain of RNA
polymerase IIO. J Biol Chem. 273:27593–27601. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Licciardo P, Amente S, Ruggiero L, et al:
The FCP1 phosphatase interacts with RNA polymerase II and with
MEP50 a component of the methylosome complex involved in the
assembly of snRNP. Nucleic Acids Res. 31:999–1005. 2003. View Article : Google Scholar : PubMed/NCBI
|