Introduction
Phosphorylation/dephosphorylation by protein kinases
is one of the most important mechanisms mediating signal
transduction (1). The
mitogen-activated protein kinase (MAPK) cascade is essential in the
transduction of signals from the extracellular space to the nucleus
(2). Four signaling pathways of
the MAPK family have been identified in eukaryotic cells as
follows: (i) The extracellular signal-regulated protein kinase
(ERK); (ii) c-Jun N-terminal kinase (JNK)/stress-activated protein
kinase (SAPK); (iii) ERK5/big MAP kinase (BMK1); and (iv) p38MAPK
pathways (3). p38MAPK is a kinase
induced by stress signals and can also be referred to as p38αMAPK
(4). Other isoforms exist in the
p38MAPK family, including p38β, p38γ (ERK6/SAPK3) and p38δ
(5). Among these isoforms,
p38αMAPK has been widely demonstrated to function in a number of
cellular functions, including differentiation, cell motility,
developmental processes and survival (6,7).
There are conflicting views on whether p38MAPK is a positive or
negative regulator of apoptosis (8,9).
Tumor necrosis factor-α (TNF-α), a cytokine with
high immunological competence, is implicated in cell proliferation
and apoptosis (10). TNF-α has
been demonstrated to serve a paradoxical role in the evolution and
treatment of cancer. Certain studies have demonstrated that TNF-α
inhibits cell viability and induces apoptosis by binding to its
specific receptors on the tumor cell membrane (11–13),
or by activating signaling pathways including ERK, JNK, p38MAPK,
nuclear factor-κB (NF-κB) and caspase cascades (14). In rat fetal brown adipocytes,
p38MAPK mediates TNF-α-induced apoptosis (15). However, studies have reported
contradictory results; in murine fibroblasts, TNF-α-induced
cytotoxicity was enhanced by p38MAPK inhibition (16), whilst in LNCaP prostatic cancer
cells, p38MAPK was demonstrated to protect against TNF-α-induced
apoptosis (17). However, little
is known regarding the p38MAPK signaling involved in TNF-α-induced
apoptosis in glioma cells.
In the current study, it was hypothesized that the
activation of p38MAPK mediates TNF-α-induced apoptosis in glioma C6
cells, suggesting p38MAPK as a potential target for glioma
therapy.
Materials and methods
Cell culture
Rat glioma C6 cells were obtained from the American
Type Culture Collection (Manassas, VA, USA) and maintained in RPMI
1640 medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with
10% fetal bovine serum (Gibco-BRL, Grand Island, NY, USA) and
antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin;
Gibco-BRL). The cell lines were maintained at 37°C in a humidified
air atmosphere with 5% CO2.
MTT assay
To detect cell survival subsequent to treatment, a
standard cell proliferation assay was performed (MTT reduction
assay) as in a previous study (18). Cells were seeded at a density of
1×104 cells/well in 96-well plates and incubated with
rat recombinant TNF-α (specific activity 2×107 U/mg) for
24 h (TNF-α concentrations: 0, 1×104, 1×105,
2×105 and 5×105 U/L) (Sigma-Aldrich). MTT (20
μl; Amresco, Solon, OH, USA) was added to each well and the cells
were incubated at 37°C for 4 h. Subsequently, 150 μl dimethyl
sulfoxide (Sigma-Aldrich) was added. The optical density (OD) was
then measured at a wavelength of 570 nm using a microplate reader
(Multiskan MK3; Thermo Fisher Scientific, Vantaa, Finland). The
inhibitory rate of tumor cell proliferation (%) was measured with
the following formula: (OD value of the control group − OD value of
the test group) × 100/OD value of the control group. Based on the
IC50 of TNF-α in C6 cells, one concentration of TNF-α
was selected for the experiments that followed.
Transmission electron microscopy
(TEM)
The morphology of apoptotic cells was measured using
an H-800 transmission electron microscope (Hitachi, Ltd., Tokyo,
Japan). Subsequent to treatment, the C6 cells were fixed with
glutaraldehyde (Sigma-Aldrich) for 1 h, washed twice with
phosphate-buffered saline (PBS; Gibco-BRL) and then suspended with
osmic acid (Sigma-Aldrich). Following a 1-h resting period, the
cells were gradually dehydrated with acetone (Sigma-Aldrich) and
embedded in resin (PRIMASET PT-30; Lonza Japan Ltd, Chiba, Japan).
The cells were then placed onto slides and observed using TEM
(JEM-2000EX; JEOL, Ltd, Tokyo, Japan).
Cell cycle analysis
Apoptosis was analyzed by flow cytometry. Subsequent
to treatment, 1×106 glioma C6 cells were prepared into a
cell suspension by trypsinization (Sigma-Aldrich), and washed twice
with ice-cold PBS. Cells were fixed with 70% ethanol
(Sigma-Aldrich) at 4°C overnight, then were treated with propidium
iodide (Sigma-Aldrich) and RNase A (Nacalai Tesque, Inc., Kyoto,
Japan) at 37°C for 40 min. Subsequently, samples were analyzed with
a FACSCalibur flow cytometry system (BD Biosciences, San Jose, CA,
USA). The sub-G1 population, representing apoptotic
cells, was scored using the hypodiploid DNA content, which was
quantified using ModFit LT software, version 3.2 (Verity Software
House, Inc., Topsham, ME, USA); the percentages of DNA
fragmentation reflecting apoptotic cells were determined by
measuring the fraction of nuclei containing a hypodiploid DNA
(sub-G1 peak), which based on >5,000 cells analyzed by flow
cytometry.
Western blot analysis
For the detection of the p38MAPK (1:500; sc-166357;
mouse anti-rat monoclonal antibodies), phosphorylated-p38MAPK
(1:500; sc-166182; mouse anti-rat polyclonal antibodies), JNK
(1:500; sc-7345; mouse anti-rat monoclonal antibodies) and
phosphorylated-JNK (1:500; sc-6254; mouse anti-rat monoclonal
antibodies) proteins (Santa Cruz Biotechnology, Inc., Dallas, TX,
USA), the whole-cell extracts were lysed in lysis buffer
(Sigma-Aldrich) and then clarified by centrifugation at 12,000 × g
for 10 min at 4°C. The volume of protein was normalized by the
Bradford method, as previously described (19). Equal amounts of total protein were
loaded onto 10–15% SDS-PAGE gels (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) for electrophoresis (Bio-Rad Laboratories, Inc.)
and the separated proteins were subsequently electrotransferred
onto polyvinylidene fluoride membranes (Millipore, Bedford, MA,
USA). The membranes were then blocked with 5% nonfat milk in PBS
with Tween-20 (Sigma-Aldrich), and probed with various primary
antibodies (as described above) overnight at 4°C, then secondary
antibodies (goat anti-mouse Immunoglobulin G-horseradish
peroxidase; 1:2,000; sc-2005; Santa Cruz Biotechnology, Inc.) for 1
h. The membranes were subsequently washed and the bands were
examined using a ChemiDoc XRS system (Bio-Rad Laboratories, Inc.)
with the enhanced chemiluminescence (ECL) western blot detection
system (Amersham Life Science, Arlington Heights, IL, USA). The
densities of the bands were determined by densitometry using
Quantity One 4.5 software (Bio-Rad Laboratories, Inc.).
Inhibitor assay
To investigate whether MAPK influences TNF-α-induced
apoptosis, C6 cells were also treated with TNF-α in the presence of
a specific inhibitor of p38MAPK (SB202190) or JNK inhibitor
(SP600125) (10 μmol/L; Calbiochem, San Diego, CA, USA) for 2 h.
TNF-α was then added for the indicated experimental groups.
Gene transfection assay
Transient transfection was carried out using
Lipofectin (Invitrogen Life Technologies, Carlsbad, CA, USA)
following the manufacturer’s instructions. During the logarithmic
growth phase, C6 cells were transferred into a 6-well plate at a
density of 2×108cells/well and were incubated for 24 h.
C6 cells were washed with Serum-free medium (0.8 ml; Gibco-BRL)
prior to the addition of 2 μg pCMV5-p38MAPK (Promega Corp.) and 10
μl Lipofectin, which were dissolved in 100 μl serum-free medium,
and then rested for 30 min at room temperature. Following 12-h
incubation, 1 ml medium with 15% serum was added and then incubated
for 24 h. The control was transfected with pCMV5 plasmid (Addgene,
Inc., Cambridge, MA, USA) and non-transfected C6 cells. Subsequent
to transfection, certain cells were fixed for TEM observation. The
cell cycle distribution was analyzed by flow cytometry, and the
expression of p38MAPK was detected by western blot analysis as
described in the above methods.
ELISA
The rat soluble TNF-α (sTNF-α) ELISA kit (Invitrogen
Life Technologies) was used to perform the ELISA in accordance with
the manufacturer’s instructions. Following p38MAPK transfection,
the culture supernatants were collected. The coating TNF-α goat
anti-rat polyclonal antibody (1:100; sc-1349; Santa Cruz
Biotechnology, Inc.) was added into an ELISA plate (Invitrogen Life
Technologies) at 100 μl/well for 48 h at 4°C. Subsequently, the
plate was washed with PBS containing 0.05% Tween-20 three times. A
total of 100 μl/well of the detected sample, negative control and
standard sample were added at proportional dilutions with PBS
(1:1), seperately. Following incubation at 37°C for 1 h, the plate
was washed three times. A total of 100 μl/well of the horseradish
peroxidase-labeled donkey anti-goat monoclonal antibody (1:100;
sc-2020; Santa Cruz Biotechnology, Inc.) was added, then following
incubation at 37°C for 1 h, the plate was washed three times with
PBS containing 1% bovine serum albumin (BSA; Sigma-Aldrich). A
total of 100 μl/well
2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)-diammonium
salt (ABTS; Sigma-Aldrich) was added and 15 min later, the OD was
detected by the ELISA reader (MR 5000; Dynatech Laboratories,
Chantilly, VA, USA) at a wavelength of 410 nm. The standard curve
was drawn and the concentrations of sTNF-α in the sample were
calculated.
Flow cytometry for membrane TNF-α and
membrane TNF receptor I (TNFRI) detection
Cells in each group were prepared into a cell
suspension using trypsin and washed with PBS containing 1% BSA; the
final concentration was 1×106/ml. A total of 2 μl
rabbit-anti-rat membrane TNF-α polyclonal antibody (1:1,000; 3707S;
Cell Signaling Technology, Inc., Beverly, MA, USA) or TNFRI
polyclonal antibody (1:1,000; 13377S; Cell Signaling Technology,
Inc.) was added into 0.5 ml cell suspension. The cell suspension
was shaken every 5 min for 30 min, at 37°C. Subsequent to washing
with PBS containing 1% BSA, goat-anti-rabbit monoclonal
immunoglobulin G (1:2,000; 7074S; Cell Signaling Technology)
labeled by fluorescein isothiocyanate was added and washed with PBS
containing 1% BSA three times. Subsequent to the addition of 1 ml
4% paraformaldehyde (Sigma-Aldrich), the cells were observed by
fluorescence microscopy (DP90; Olympus, Tokyo, Japan) and detected
by flow cytometry using a FACS LSR II system (Becton Dickinson, San
Jose, CA, USA).
Statistical analysis
All data are presented as the mean ± standard
deviation of at least three independent experiments. Statistical
differences between the means were analyzed by the
independent-samples t-test. Rates were compared using the
χ2 test. P<0.05 was considered to indicate a
statistically significant difference. All statistical analyses were
performed with SPSS software, version 17.0 (SPSS, Inc., Chicago,
IL, USA).
Results
TNF-α inhibits the proliferation of
glioma C6 cells
The MTT assay was used to detect cell viability of
glioma C6 cells. Subsequent to treatment with TNF-α, the
proliferation of C6 cells was inhibited in a dose-dependent manner.
The inhibitory rates of different concentrations of TNF-α on C6
cells are described in Table I.
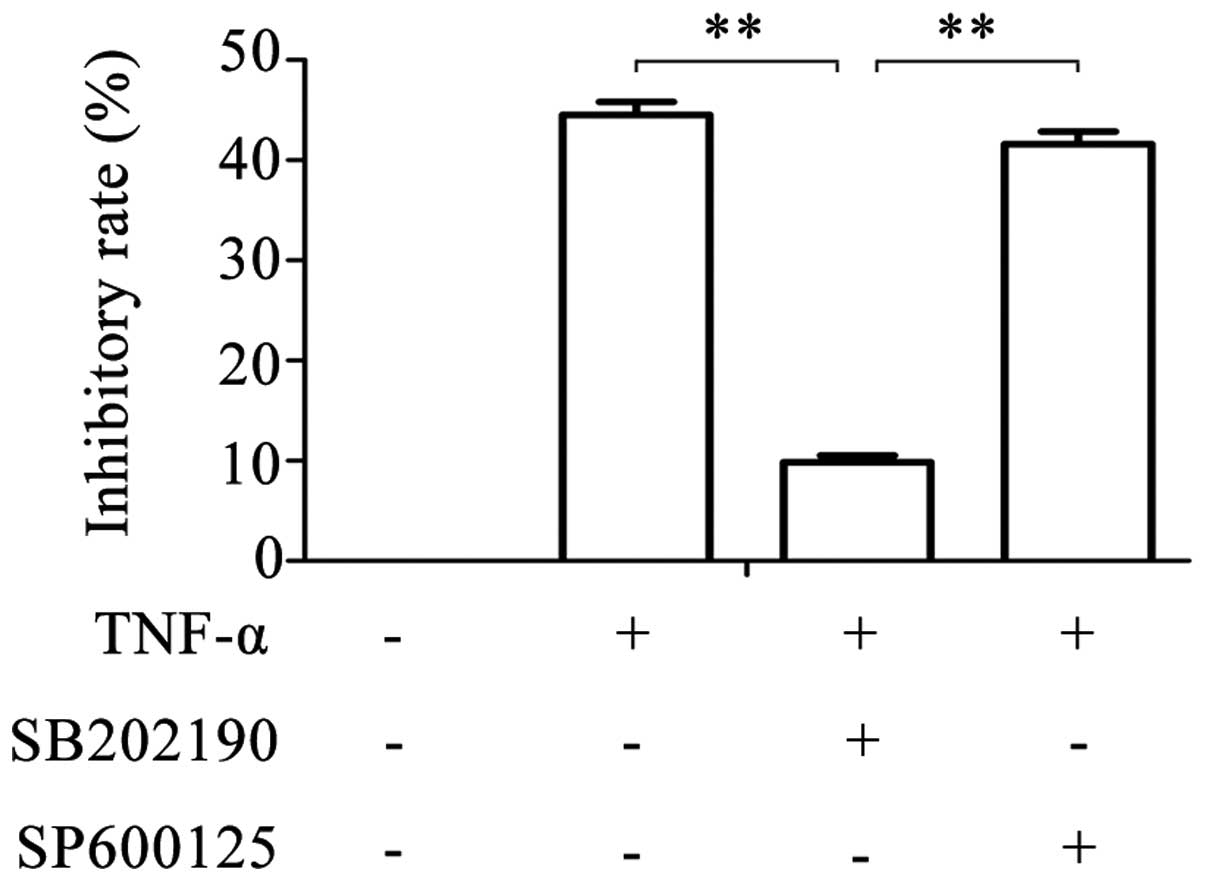
The IC50 of TNF-α in C6 cells was 2.53×105
U/L, and this concentration was used for the subsequent
experiments. Following treatment of glioma cells with SB202190 and
TNF-α simultaneously, the inhibitory rate reduced to 9.849±0.675%
(Fig. 1). However, following
simultaneous treatment with SP600125 and TNF-α, the inhibitory rate
was similar to that of TNF-α treatment alone (41.615±1.236% vs.
44.518±1.305%). These results suggest that TNF-α is
antiproliferative in a dose-dependent manner in glioma cells, and
that this effect of TNF-α can be partly blocked by the inhibition
of p38MAPK with SB202190, but not by inhibition of JNK with
SP600125.
 | Table IInhibitory rates of TNF-α on the
proliferation of glioma cells. |
Table I
Inhibitory rates of TNF-α on the
proliferation of glioma cells.
| TNF-α (U/l) | OD570 (mean ±
SD) | Inhibitory rate
(%) |
|---|
| 0 | 0.995±0.004 | 0 |
|
1×104 | 0.962±0.018 | 3.316±0.603 |
|
1×105 | 0.645±0.031 | 35.180±1.276 |
|
2×105 | 0.552±0.030 | 44.518±1.305 |
|
5×105 | 0.378±0.009 | 62.012±2.229 |
TNF-α-induces apoptosis in rat C6
cells
To investigate the mechanisms associated with a
TNF-α-induced reduction in cell proliferation, the effects of TNF-α
treatment on cell cycle arrest and apoptosis were examined.
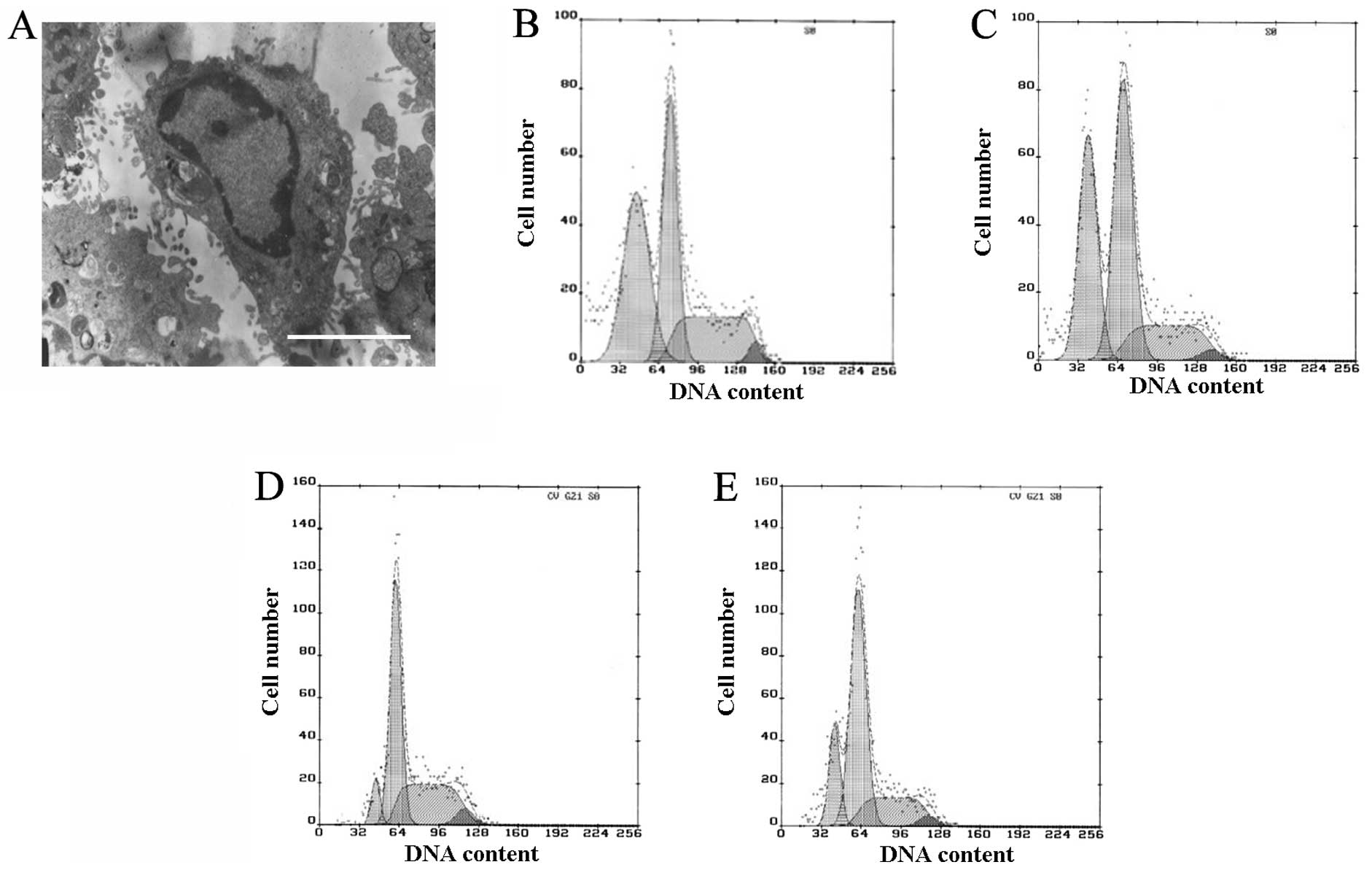
Apoptosis was observed by TEM and quantified by flow cytometry. In
the TNF-α and SP600125-treated groups, a number of the cells
exhibited clear apoptotic characteristics, including pyknosis,
chromatin condensation and formation of apoptotic bodies, when
observed by TEM (Fig. 2A). Few
apoptotic cells were observed in the control and SB202190-treated
groups, whereas in the TNF-α- and SP600125-treated groups, the
proportion of cells in the G1 phase was increased and
the proportions in the S and G2 phases were reduced, and
clear apoptotic peaks emerged (Fig
2B–E). The apoptotic rates in the TNF-α- and SP600125-treated
groups were 37.5 and 34.1%, respectively (Fig. 2B and C). There was a smaller
apoptotic peak in the control and SB202190-treated groups, in which
the apoptotic rates were 7 and 16.9%, respectively (Fig. 2D and E). The apoptotic rates of the
TNF-α- and SP600125-treated groups were significantly different
from that of the control or SB202190-treated group (P<0.01; data
not shown). The results of the current study demonstrate that
TNF-α-induced apoptosis may be partly blocked by the inhibition of
p38MAPK, but not by inhibition of JNK.
p38MAPK activation is involved in
TNF-α-induced apoptosis
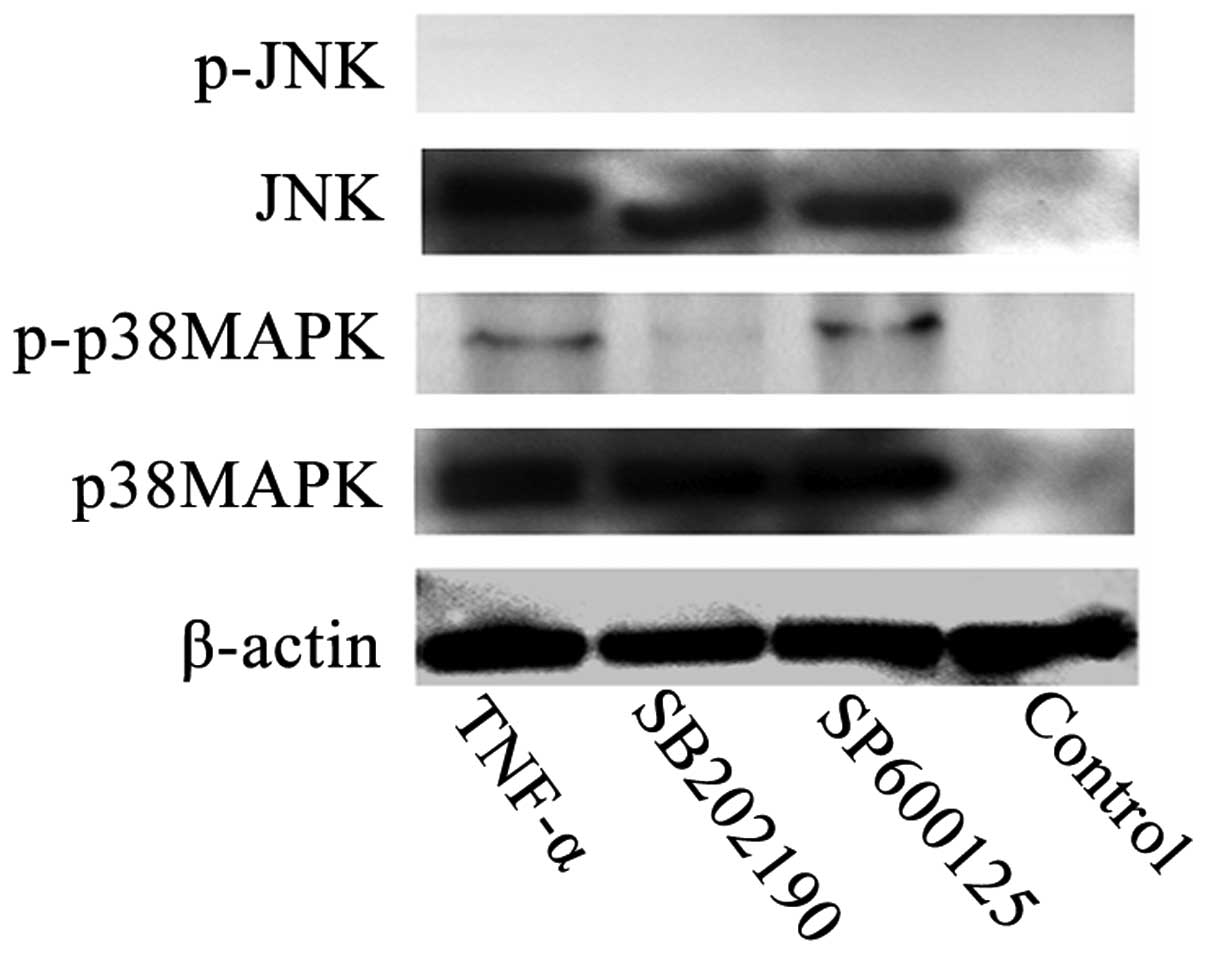
To investigate the potential involvement of MAPK in
TNF-α-induced apoptosis, the activation states of p38MAPK and JNK
were investigated using western blot analysis with antibodies
specific to the phosphorylated forms of these kinases. Subsequent
to 2×105 U/L TNF-α treatment, clear activation of
phosphorylated-p38MAPK was observed (Fig. 3). In contrast, induction of JNK
phosphorylation was not observed in any group. In addition, no
phosphorylated-p38MAPK band was detected in the SB202190-treated or
control group (Fig. 3). These data
suggest that p38MAPK phosphorylation, but not JNK phosphorylation,
may be involved in the mediation of TNF-α-induced apoptosis.
p38MAPK gene transfection induces
apoptosis
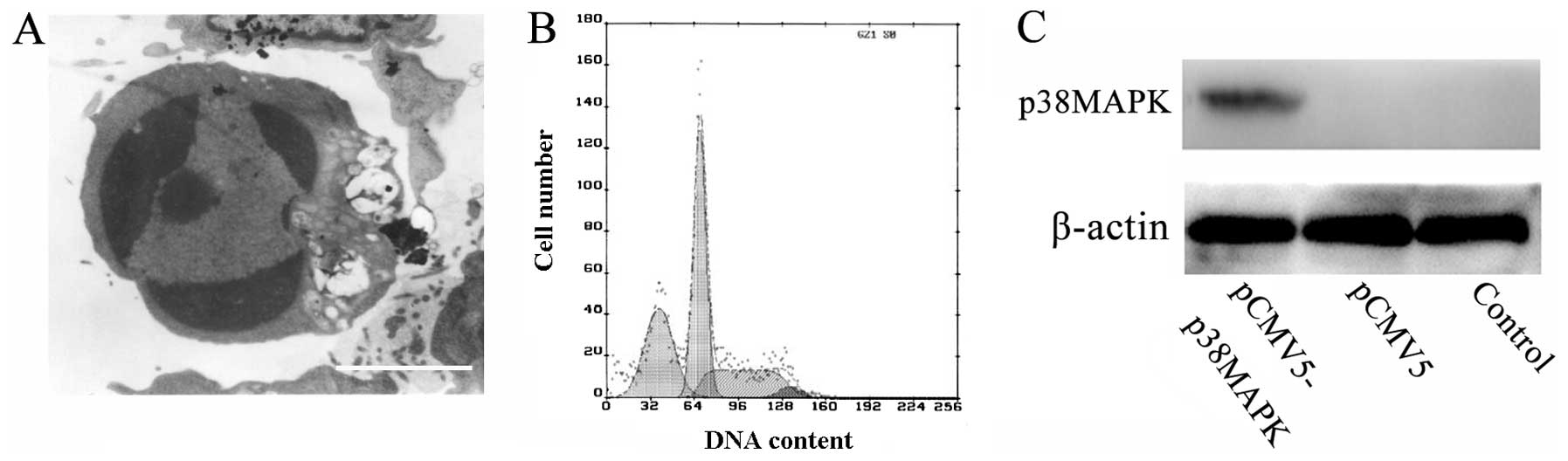
To investigate the role of p38MAPK in TNF-α-induced
apoptosis, transient transfection was carried out using Lipofectin.
During the 24-h pCMV5-p38MAPK transfection, the asteroid-shaped
cells became bipolar or round in shape, with reduced sizes and
increased intracellular bubbles. After 24 h, a number of the dead
cells floated in the medium. There were no distinct alterations in
the non-transfected and pCMV5 groups. In addition, a number of
cells demonstrated clear apoptotic alterations, including pyknosis,
chromatin condensation and formation of apoptotic bodies (Fig. 4A). Cell cycle analysis identified a
clear apoptotic peak in the pCMV5-p38MAPK group and the apoptotic
index was 31.2% (Fig. 4B). Western
blot analysis demonstrated that a distinct immunoreactive band at
38 kDa was present in the pCMV5-p38MAPK group. No band was present
in the non-transfected or pCMV5 groups (Fig. 4C). Additionally, the sTNF-α
concentration was analyzed subsequent to gene transfection. The
results demonstrated that the concentration of sTNF-α in the
culture supernatant of the pCMV5-p38MAPK group increased and
reached 43.4 pg/ml following treatment for 24 h, but there was no
clear alteration in the pCMV5 and non-transfected groups (data not
shown).
Expression of membrane TNF-α and TNFRI
following gene transfection
The expression rates of membrane TNF-α in the pCMV5,
pCMV5-p38MAPK and control (non-transfected) groups were
4.079±0.532, 6.218±0.601 and 3.845±0.482%, respectively, and were
not significantly different. The expression rates of membrane TNFRI
in the pCMV5, pCMV5-p38MAPK and control groups were 5.422±0.551,
14.906±0.627 and 3.633±0.404%, respectively. Out of these, the
level in the pCMV5-p38MAPK group was significantly higher than that
of the other two groups (P<0.01; Fig. 5).
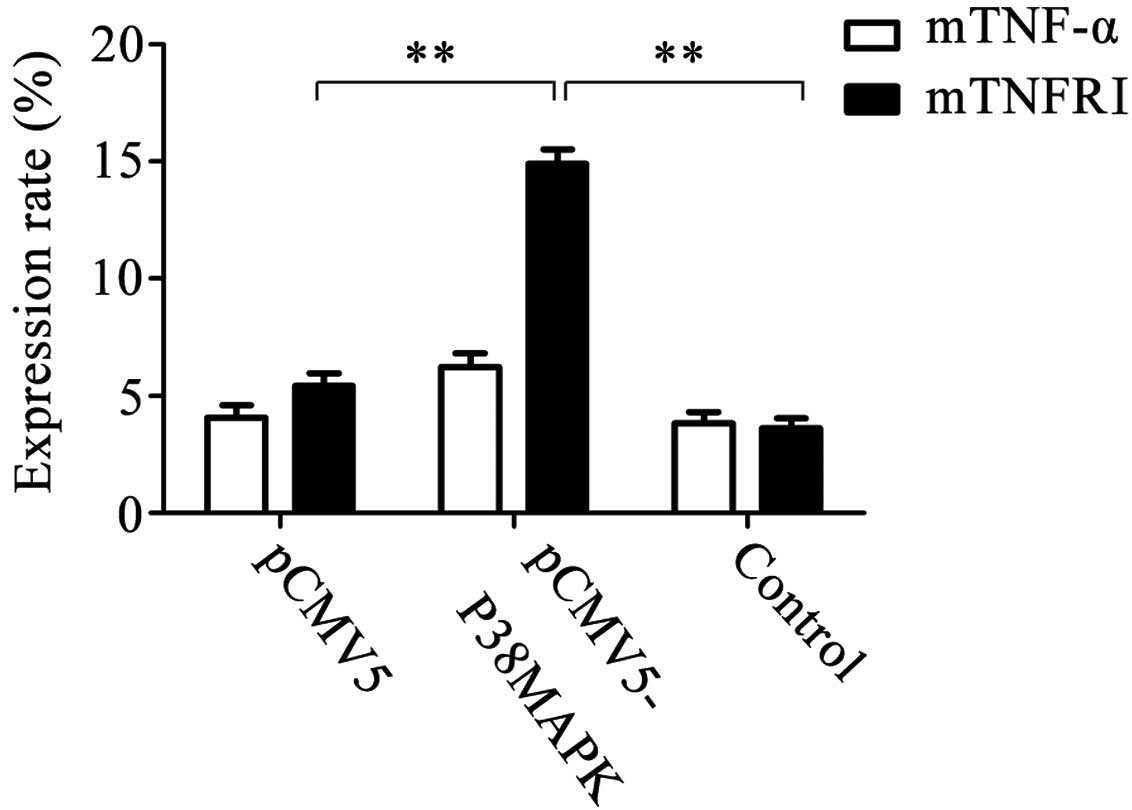 | Figure 5Expression of membrane TNF-α and
membrane TNFRI subsequent to gene transfection. The results
demonstrated that the expression rates of membrane TNF-α in the
pCMV5 group, pCMV5-p38MAPK group and non-transfected group were
4.079±0.532, 6.218±0.601 and 3.845±0.482%, respectively. There were
no significant differences between them. The expression rates of
membrane TNFRI in the pCMV5 group, pCMV5-p38MAPK group and
non-transfected group were 5.422±0.551, 14.906±0.627 and
3.633±0.404%, respectively. Among them, the expression in the
pCMV5-p38MAPK group was significantly higher than that in the other
two groups **P<0.01, n=5. TNF-α, tumor necrosis
factor-α; TNFRI, TNF receptor I. |
Discussion
The current study demonstrated that TNF-α inhibits
proliferation and induces apoptosis in glioma cells. Apoptosis, a
biological process characterized by condensation of the nucleus and
a distinctive pattern of chromosomal DNA fragmentation, is
established to be essential for the development and maintenance of
homeostasis during cell growth and the elimination of damaged cells
in multicellular organisms (20).
Disruption to apoptotic regulation is closely associated with the
generation, development and prognosis of tumors. TNF-α regulates
immune function and cytotoxicity in tumor cells by binding to its
receptor, and the induction of apoptosis and cell cycle inhibition
has been suggested as a potential mechanism for its anticancer
effects. However, the sensitivity of different types of tumor cells
to TNF-α varies, and little research has been undertaken regarding
glioma cells. In the present study, the proliferation of C6 cells
was inhibited by TNF-α through the induction of apoptosis. The
mechanism that underlies TNF-α-induced apoptosis remains unclear.
Yin et al (21)
demonstrated that TNF-α may activate wild-type p53 protein by
suppressing mutant p53, inducing p53-dependent apoptosis in glioma
cells. However, studies have reported contradictory results that
suggest that p38MAPK has an anti-apoptotic effect in TNF-α-induced
apoptosis (16,17).
The findings of the current study demonstrated that
p38MAPK activation was observed to partially mediate TNF-α-induced
apoptosis of glioma cells. In 1994, Han et al (22) cloned p38MAPK from the murine liver
cDNA library, then detected the expression of p38MAPK mRNA in
murine macrophages (T and B cells). p38MAPK is activated by
ultraviolet light, hyperosmolarity, arsenate, heat shock,
H2O2, cytokines (such as IL-1 and TNF-α) and
physiological stress (23).
Following translocation from the nucleus to the cytoplasm, it
initiates the activity of corresponding transcription factors. It
has been reported that various transcription factors, including
ATF2, MEF2C, CHOP10 and SAP1, are the physiological substrates of
p38MAPK (24). p38MAPK acts in the
pathophysiological processes of cell differentiation, development
and regulation of apoptosis. In previous studies, p38MAPK
activation was identified to induce apoptosis in non-tumor cells,
such as nerve cells (7), fetal
brown adipocytes (15) and tumor
cells (25–27). However, a number of studies have
identified p38MAPK to be independent of apoptosis (28), or even to inhibit it (29,30).
The involvement of p38MAPK in TNF-α-induced
apoptosis remains unclear and previous evidence is contradictory.
In human cervix carcinoma cells, p38MAPK activation may be a key
upstream signal of TNF-α-induced apoptosis and attenuation of the
p38MAPK pathway by overexpression of DDB2 (a DNA repair protein)
may be responsible for acquired TNF-α resistance (31). ASK1, a MAPKKK activated in cells
treated with TNF-α, activates MKK3/MAPKK6 (or MKK6) and p38MAPK in
turn, thus inducing apoptosis (32). However, another study demonstrated
that ASK1 mediates anti-apoptotic signals (33). Insulin and insulin-like growth
factor-I (IGF-I) protect HT29-D4 colon carcinoma cells from
IFN-γ/TNF-α-induced apoptosis. The anti-apoptotic function of IGF-I
is based on the enhancement of the survival pathways initiated by
TNF-α and is mediated by p38MAPK, ERK and NF-κB, which act together
to suppress proapoptotic signals (9). In L929 cells that overexpress SSI-1
and have been treated with TNF-α, the activation of p38MAPK was
observed to be sustained, and the cells were resistant to
TNF-α-induced apoptosis (34). In
the current study, p38MAPK phosphorylation, but not JNK
phosphorylation, was detected in the TNF-α-treated C6 cells.
Subsequent to treatment with SB202190 and TNF-α, no positive
p38MAPK signal was detected in the western blot analysis, and the
apoptosis was higher than in the SB202190 and control groups. These
data suggested that the p38MAPK, but not the JNK signaling pathway,
is involved in TNF-α-induced apoptosis.
The current study demonstrated that p38MAPK
transfection enhances apoptosis. To further investigate the role of
p38MAPK in TNF-α-induced apoptosis, transient transfection was
performed using Lipofectin. Subsequent to the transfection, p38MAPK
was clearly expressed and confirmed to induce apoptosis. Activation
of p38MAPK may contribute to the pathogenesis of apoptosis via the
following mechanisms: (i) Upregulation of specific oncogenes,
including c-Myc/s-Myc, c-Fos, c-Jun and bax (35); (ii) participating in
Fas/FasL-mediated apoptosis (36);
(iii) inducing p53 phosphorylation (37); and (iv) enhancing TNF-α expression
(38). In endotoxin-activated
glial cells, Bhat et al (39) demonstrated the importance of ERK
and p38MAPK cascades in the transcriptional and
post-transcriptional regulation of iNOS and TNF-α gene expression.
Anti-TNF strategies targeting p38MAPK, NF-κB and TNF-α are being
investigated as possible methods for the attenuation of renal
ischemic injury (40). Soluble
TNFRI, one of the two forms of TNFRI (soluble and membrane type),
usually restricts the activity of TNF-α and is regarded as its
antagonist. However, sTNF-α (the functional type of TNF-α) induces
apoptosis when it binds to membrane TNFRI, which in turn binds to
the TNF receptor-associated death domain (TRADD) molecules.
TNFRI/TRADD binding results in the activation of a protein cascade
(TRAF-2, ASK-1, MEK-4 and JNK) that ends with the activation of
activator protein-1 (AP-1) (41).
In the current study, the data demonstrated that p38MAPK
transfection enhances apoptosis by the upregulation of sTNF-α and
membrane TNFRI, which inferred that p38MAPK activation may
contribute to the pathogenesis of apoptosis via these
mechanisms.
In conclusion, the current study demonstrated that
activation of p38MAPK, but not JNK, mediates TNF-α-induced
apoptosis in glioma C6 cells. This suggests that the enhancement of
p38MAPK activity can inhibit the progression of glioma cells.
However, the MAPK signaling pathway is complex, thus it is
necessary to further investigate the role of the upstream or
downstream kinases in the p38MAPK cascade of apoptosis, in addition
to other MAPKs, such as ERK1/2 or ERK5. In vivo experiments
are also required to verify the results of the current study.
References
|
1
|
Sawyer TK, Shakespeare WC, Wang Y, et al:
Protein phosphorylation and signal transduction modulation:
chemistry perspectives for small-molecule drug discovery. Med Chem.
1:293–319. 2005. View Article : Google Scholar
|
|
2
|
Brown MD and Sacks DB: Protein scaffolds
in MAP kinase signalling. Cell Signal. 21:462–469. 2009. View Article : Google Scholar :
|
|
3
|
Rana A, Rana B, Mishra R, et al: Mixed
lineage kinase-c-Jun N-terminal kinase axis: a potential
therapeutic target in cancer. Genes Cancer. 4:334–341. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fischer S, Koeberle SC and Laufer SA: p38α
mitogen-activated protein kinase inhibitors, a patent review
(2005–2011). Expert Opin Ther Pat. 21:1843–1866. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cohen P: The search for physiological
substrates of MAP and SAP kinases in mammalian cells. Trends Cell
Biol. 7:353–361. 1997. View Article : Google Scholar
|
|
6
|
Borders AS, de Almeida L, Van Eldik LJ and
Watterson DM: The p38alpha mitogen-activated protein kinase as a
central nervous system drug discovery target. BMC Neurosci. 9(Suppl
2): S122008. View Article : Google Scholar
|
|
7
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zechner D, Craig R, Hanford DS, McDonough
PM, Sabbadini RA and Glembotski CC: MKK6 activates myocardial cell
NF-kappaB and inhibits apoptosis in a p38 mitogen-activated protein
kinase-dependent manner. J Biol Chem. 273:8232–8239. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Remacle-Bonnet MM, Garrouste FL, Heller S,
André F, Marvaldi JL and Pommier GJ: Insulin-like growth factor-I
protects colon cancer cells from death factor-induced apoptosis by
potentiating tumor necrosis factor alpha-induced mitogen-activated
protein kinase and nuclear factor kappaB signaling pathways. Cancer
Res. 60:2007–2017. 2000.PubMed/NCBI
|
|
10
|
Zidi I, Mestiri S, Bartegi A and Amor NB:
TNF-alpha and its inhibitors in cancer. Med Oncol. 27:185–198.
2010. View Article : Google Scholar
|
|
11
|
Szlosarek PW and Balkwill FR: Tumour
necrosis factor alpha: a potential target for the therapy of solid
tumours. Lancet Oncol. 4:565–573. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gray-Schopfer VC, Karasarides M, Hayward R
and Marais R: Tumor necrosis factor-alpha blocks apoptosis in
melanoma cells when BRAF signaling isinhibited. Cancer Res.
67:122–129. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sun SY, Yue P, Hong WK and Lotan R:
Augmentation of tumor necrosis factor-related apoptosis-inducing
ligand (TRAIL)-induced apoptosis by the synthetic retinoid
6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphthalene carboxylic acid
(CD437) through up-regulation of TRAIL receptors in human lung
cancer cells. Cancer Res. 60:7149–7155. 2000.
|
|
14
|
Barbin G, Roisin M and Zalc B: Tumor
necrosis factor α activates the phosphorylation of ERK, SAPK/JNK,
and P38 kinase in primary cultures of neurons. Neurochem Res.
26:107–112. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Valladares A, Alvarez AM, Ventura JJ,
Roncero C, Benito M and Porras A: p38 mitogen-activated protein
kinase mediates tumor necrosis factor-alpha-induced apoptosis in
rat fetal brown adipocytes. Endocrinology. 141:4383–4395.
2000.PubMed/NCBI
|
|
16
|
Lüschen S, Scherer G, Ussat S, Ungefroren
H and Adam-Klages S: Inhibition of p38 mitogen-activated protein
kinase reduces TNF-induced activation of NF-kappaB, elicits caspase
activity, and enhances cytotoxicity. Exp Cell Res. 293:196–206.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ricote M, García-Tuñón I, Fraile B,
Fernández C, Aller P, Paniagua R and Royuela M: P38 MAPK protects
against TNF-alpha-provoked apoptosis in LNCaP prostatic cancer
cells. Apoptosis. 11:1969–1975. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang X, Yao J, Luo Y, Han Y, Wang Z and Du
L: P38 MAP kinase mediates apoptosis after genipin treatment in
non-small-cell lung cancer H1299 cells via a mitochondrial
apoptotic cascade. J Pharmacol Sci. 121:272–281. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bradford MM: A rapid and sensitive for the
quantitation of microgram quantitites of protein utilizing the
principle of protein-dye binding. Analytical Biochemistry.
72:248–254. 1976. View Article : Google Scholar
|
|
20
|
Allegra A, Alonci A, Campo S, Penna G,
Petrungaro A, Gerace D and Musolino C: Circulating microRNAs: new
biomarkers in diagnosis, prognosis and treatment of cancer
(review). Int J Oncol. 41:1897–1912. 2012.PubMed/NCBI
|
|
21
|
Yin D, Kondo S, Barnett GH, Morimura T and
Takeuchi J: Tumor necrosis factor-alpha induces p53-dependent
apoptosis in rat glioma cells. Neurosurgery. 37:758–763. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Han J, Lee JD, Bibbs L and Ulevitch RJ: A
MAP kinase targeted by endotoxin and hyperosmolarity in mammalian
cells. Science. 265:808–811. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ashwell JD: The many paths to p38
mitogen-activated protein kinase activation in the immune system.
Nat Rev Immunol. 6:532–540. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cuadrado A and Nebreda AR: Mechanisms and
functions of p38 MAPK signalling. Biochem J. 429:403–17. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ishii A, Furusho M and Bansal R: Sustained
activation of ERK1/2 MAPK in oligodendrocytes and schwann cells
enhances myelin growth and stimulates oligodendrocyte progenitor
expansion. J Neurosci. 33:175–186. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Park MT, Choi JA, Kim MJ, et al:
Suppression of extracellular signal-related kinase and activation
of p38 MAPK are two critical events leading to caspase-8- and
mitochondria-mediated cell death in phytosphingosine-treated human
cancer cells. J Biol Chem. 278:50624–50634. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mandal C, Dutta A, Mallick A, Chandra S,
Misra L, Sangwan RS and Mandal C: Withaferin A induces apoptosis by
activating p38 mitogen-activated kinase signaling cascade in
leukemic cells of lymphoid and myeloid origin through mitochondrial
death cascade. Apoptosis. 13:1450–1464. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ozaki I, Tani E, Ikemoto H, Kitagawa H and
Fujikawa H: Activation of stress-activated protein kinase/c-Jun
NH2-terminal kinase and p38 kinase in calphostin C-induced
apoptosis requires caspase-3-like proteases but is dispensable for
cell death. J Biol Chem. 274:5310–5317. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kanagasabai R, Karthikeyan K, Vedam K,
Qien W, Zhu Q and Ilangovan G: Hsp27 protects adenocarcinoma cells
from UV-induced apoptosis by Akt and p21-dependent pathways of
survival. Mol Cancer Res. 8:1399–1412. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Park JG, Yuk Y, Rhim H, Yi SY and Yoo YS:
Role of p38 MAPK in the regulation of apoptosis signaling induced
by TNF-alpha in differentiated PC12 cells. J Biochem Mol Biol.
35:267–272. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sun CL and Chao CC: Potential attenuation
of p38 signaling by DDB2 as a factor in acquired TNF resistance.
Int J Cancer. 115:383–387. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lin CY, Chang SL, Fong YC, Hsu CJ and Tang
CH: Apoptosis signal-regulating kinase 1 is involved in
brain-derived neurotrophic factor (BDNF)-enhanced cell motility and
matrix metalloproteinase 1 expression in human chondrosarcoma
cells. Int J Mol Sci. 14:15459–15478. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Won M, Park KA, Byun HS, et al: Novel
anti-apoptotic mechanism of A20 through targeting ASK1 to suppress
TNF-induced JNK activation. Cell Death Differ. 17:1830–1841. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Morita Y, Naka T, Kawazoe Y, et al:
Signals transducers and activators of transcription (STAT)-induced
STAT inhibitor-1 (SSI-1)/suppressor of cytokine signaling-1
(SOCS-1) suppresses tumor necrosis factor alpha-induced cell death
in fibroblasts. Proc Natl Acad Sci USA. 97:5405–5410. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Noguchi K, Yamana H, Kitanaka C, Mochizuki
T, Kokubu A and Kuchino Y: Differential role of the JNK and p38
MAPK pathway in c-Myc- and s-Myc-mediated apoptosis. Biochem
Biophys Res Commun. 267:221–227. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cho SD, Ahn NS, Jung JW, et al: Critical
role of the c-JunNH2-terminal kinase and p38 mitogen-activated
protein kinase pathways on sodium butyrate-induced apoptosis in
DU145 human prostate cancer cells. Eur J Cancer Prev. 15:57–63.
2006. View Article : Google Scholar
|
|
37
|
Van Laethem A, Van Kelst S, Lippens S, et
al: Activation of p38 MAPK is required for Bax translocation to
mitochondria, cytochrome c release and apoptosis induced by UVB
irradiation in human keratinocytes. FASEB J. 18:1946–1948.
2004.PubMed/NCBI
|
|
38
|
Keller D, Zeng X, Li X, et al: The p38MAPK
inhibitor SB203580 alleviates ultraviolet-induced phosphorylation
at serine 389 but not serine 15 and activation of p53. Biochem
Biophys Res Commun. 261:464–471. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bhat NR, Zhang PS, Lee JC and Hogan EL:
Extracellular signal-regulated kinase and p38 subgroups of
mitogen-activated protein kinases regulate inducible nitric oxide
synthase and tumor necrosis factor-alpha gene expression in
endotoxin-stimulated primary glial cultures. J Neurosci.
18:1633–1641. 1998.PubMed/NCBI
|
|
40
|
Donnahoo KK, Shames BD, Harken AH and
Meldrum DR: Review article: the role of tumor necrosis factor in
renal ischemia-reperfusion injury. J Urol. 162:196–203. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ichijo H, Nishida E, Irie K, et al:
Induction of apoptosis by ASK1, a mammalian MAPKKK that activates
SAPK/JNK and p38 signaling pathways. Science. 275:90–94. 1997.
View Article : Google Scholar : PubMed/NCBI
|