Introduction
Chronic kidney disease induces irreversible kidney
damage, and progressive renal fibrosis is frequently observed
regardless of the underlying cause of the disease (1). The major effector cells that are
associated with the development of progressive renal fibrosis are
the interstitial myofibroblasts (2). Interstitial myofibroblasts have been
proposed to originate from five potential sources, including
circulating fibrocytes, pericytes, fibroblasts, the tubular
epithelial-mesenchymal transition (EMT) and the
endothelial-mesenchymal transition (3).
Fibrocytes are bone marrow-derived mesenchymal
progenitor cells, which express hematopoietic stem cell antigens,
monocytic lineage markers and fibroblast products (4). Fibrocytes constitutively produce
extracellular matrix (ECM) components, in addition to ECM-modifying
enzymes, and are able to differentiate into myofibroblasts in
vitro and in vivo under certain micro-environmental
conditions (5). There is an
increasing body of evidence suggesting that these cells contribute
to the development of the novel myofibroblast population that is
responsible for the production of ECM during renal fibrosis
(6–11).
Erythropoietin (EPO) is a hematopoietic hormone, the
majority of which is produced by the adult kidneys. In addition to
its erythropoietic effects, EPO exerts protective effects against
acute ischemic and toxic renal injuries (12–14).
EPO also protects against interstitial injuries, including
interstitial fibrosis, in a variety of animal models of human
disease (15–18). The inhibition of EMT is one
potential mechanism underlying the anti-fibrotic effects of EPO
(16,18). However, whether the beneficial
effects of EPO in renal fibrosis are associated with the inhibition
of fibrocyte accumulation remains to be elucidated. The aim of the
present study was to determine whether recombinant human (rh)EPO
treatment was able to prevent the progression of renal fibrosis via
the attenuation of interstitial fibrocytes in a mouse model of
complete unilateral ureteral obstruction (UUO).
Materials and methods
Animals, agents and antibodies
Male C57BL6 mice (18–20 g) were purchased from the
Experimental Animal Center of Wuhan University (Wuhan, China).
Animals were housed in standard rodent cages with ad libitum
access to water and food until sacrifice. All surgical and
experimental procedures were approved by the Institutional Animal
Care and Use Committee of Wuhan University (Wuhan, China). rhEPO
was obtained from Sunshine Pharmaceutical Company (Shenyang,
China). Rabbit polyclonal anti-human collagen I (cat. no. ab34710),
Rabbit polyclonal anti-human fibronectin (cat. no. ab2413), Rabbit
polyclonal anti-human α smooth muscle actin (α-SMA; cat. no.
ab5694) and Rabbit polyclonal anti-mouse CXC chemokine ligand 16
(CXCL16; cat. no. ab119350) antibodies were purchased from Abcam
(Cambridge, UK). Rabbit Polyclonal anti-mouse CXCL12 (cat. no.
PA1-29029) was purchased from Thermo Fisher Scientific (Waltham,
MA, USA). Goat Polyclonal anti-mouse CC chemokine ligand 21 (CCL21)
antibody was purchased from R&D Systems, Inc. (cat. no. AF457;
Minneapolis, MN, USA). Rabbit polyclonal anti-human actin (cat. no.
sc-7210) and Rat monoclonal anti-mouse CD45 (cat. no. sc-21739)
antibodies were purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). Donkey monoclonal anti-rabbit or donkey
anti-goat secondary antibodies were purchased from Rockland
Immunochemicals, Inc. (Limerick, PA, USA).
Experimental protocol
Twenty-four male C57BL6 mice were randomly assigned
into four groups, each comprising six mice: i) control group (Sh);
ii) UUO plus vehicle group (U+V); iii) UUO plus 300 U/kg body
weight rhEPO (U+E1) and iv) UUO plus 1,000 U/kg body body weight
rhEPO (U+E2). UUO was performed as described previously (19). Briefly, under sodium pentobarbital
(60 mg/kg body weight; Merck, Darmstadt, Germany) anesthesia, the
left ureter was exposed and completely ligated with 4-0 sutures,
following a left abdominal incision. The sham-operation mice had
their ureters exposed and manipulated, but not ligated. For rhEPO
treatment, 300 U/kg or 1,000 U/kg body weight rhEPO was
administered intraperitoneally daily from day one to day six
following UUO. In the U+V group, vehicle (phosphate-buffered
saline) was administered intraperitoneally following a protocol
identical to the rhEPO treatment. Mice were sacrificed on the
seventh day following surgery, and the obstructed kidneys were
harvested. One third of the kidney was fixed in 4% buffered
paraformaldehyde (Boshide, Wuhan, China) and embedded in paraffin
for histological and immunohistochemical studies. One third of the
kidney was stored for flow cytometric analysis, and the remaining
kidney sections were snap-frozen in liquid nitrogen and stored at
−80°C for protein and RNA extraction.
Histological and immunohistochemical
examination
Kidney sections (5 μm) were prepared from the
paraffin-embedded tissue specimens. Total collagen was identified
using picro-sirius red (PSR) staining following a previously
described method with modifications (20). For immunohistochemical examination,
the renal sections were incubated with anti-collagen I antibody
(1:100), anti-fibronectin antibody (1:100) or anti-α-SMA antibody
(1:200). The kidney specimens were incubated with the primary
antibodies overnight at 4°C, followed by a second reaction with
anti-rabbit antibody conjugated with envision polymer (Thermo
Fisher Scientific) for 30 min. Finally, a diaminobenzidine reaction
was performed on the section using a kit (TL-015-QHD; Thermo Fisher
Scientific) and hematoxylin (Boshide) was used as the
counterstain.
Western blot analysis
Western blot analysis was performed as previousy
described (21). In brief, protein
samples were heated at 100°C for 5 min and subjected to
electrophoresis on 10 or 15% SDS gels. Proteins were
electrophoretically transferred to nitrocellulose membranes
(Millipore, Billerica, MA, USA) which were subsequently incubated
with antibodies specific for α-SMA (1:500), collagen I (1:1,000),
fibronectin (1:400), CXCL12 (1:1,000), CCL21 (1:600), CXCL16
(1:500) and β-actin (1:1,000), followed by incubation with
secondary antibody conjugated with IRDye® infrared dye
(Rockland Immunochemicals, Inc.). The signals were detected using
an Odyssey Infrared Imaging System (Li-COR Biosciences, Lincoln,
NE, USA). Actin was used as an internal control.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
qPCR, following reverse transcription was used to
assess transcript levels of collagen I and fibronectin. RNA was
extracted from frozen tissue by homogenization in
TRIzol® (Invitrogen Life Technologies, Carlsbad, CA,
USA), and 1 μg aliquots of RNA were used in a reverse transcription
reaction with M-MuLV reverse transcriptase (cat. no. K1621; Thermo
Fisher Scientific). The resulting complementary DNA was used as a
template for qPCR analysis. Primers were obtained from Sangon
Biological Engineering Technology and Services (Shanghai, China),
and the specific primers were designed as follows: Forward,
5′-TGCCGCGACCTCAAGATGTG-3′ and reverse, 5′-CACAAGGGTGCTGTAGGTGA-3′
for collagen I; forward 5′-CTTCTCCGTGGAGTTTTACCG-3′ and reverse,
5′GCTGTCAAATTGAATGGTGGTG-3′ for fibronectin and forward,
5′-GGTGAAGGTCGGTGTGAACG-3′ and reverse, 5′-CTCGCTCCTGGAAGATGGTG-3′
for GAPDH. A 20 μl sample of PCR reaction solution, which included
SYBR Green PCR Master Mix (#RR420A; TaKaRa Bio, Inc., Otsu, Japan),
was amplified according to the manufacturer’s instructions.
Quantifications were performed in duplicate on the ABI Prism 7500
Sequence Detection System (Applied Biosystems Life Technologies,
Foster City, CA, USA). The calculations of the relative change in
mRNA were performed using the ΔΔCt method.
Flow cytometry
Renal cells were isolated as previously described
with modifications (22). Briefly,
the kidneys were decapsulated, minced finely and incubated at 37°C
for 40 min in phosphate-buffered saline (Boshide) containing 0.25%
trypsin (Biyuntian, Wuhan, China). Cells were filtered through a
40-μm strainer, rinsed, centrifuged and resuspended in fluorescence
activated cell sorting buffer (Boshide). Cells (5×105)
were incubated with anti-CD45 and anti-collagen I antibodies.
Subsequently, the cells were incubated with fluorescein
isothiocyanate/phycoerythrin-labeled secondary antibodies (Santa
Cruz Biotechnology, Inc.). Certain cells were incubated with
irrelevant isotype-matched antibodies (Santa Cruz Biotechnology,
Inc.) and unstained cells were used as controls. The cut-off values
were determined according to the results of the control
experiments. Data were analyzed using BD FACSDiva™ software (BD
Biosciences, Franklin Lakes, NJ, USA).
Statistical analysis
Statistical analyses of the data were performed
using the Graphpad Prism® software package, version 5.0
(Graphpad Software, Inc., La Jolla, CA, USA) Values are expressed
as the mean ± standard deviation. Multiple group comparisons were
performed by one-way analysis of variance followed by the
Bonferroni procedure for the comparison of means. P<0.05 was
considered to indicate a statistically significant difference
between values.
Results
EPO treatment suppresses renal
tubulo-interstitial fibrosis
UUO induced an increase in collagen I expression on
the PSR-stained kidney sections, in comparison to that of the
control group on day seven. By contrast, collagen levels were
reduced in the rhEPO-treated UUO kidneys, compared with those of
the vehicle treated UUO kidneys (Fig.
1). This was verified by semi-quantitative evaluation of the
fibrotic area of the PSR-stained kidney sections. Furthermore,
compared with the control and vehicle-treated UUO kidneys,
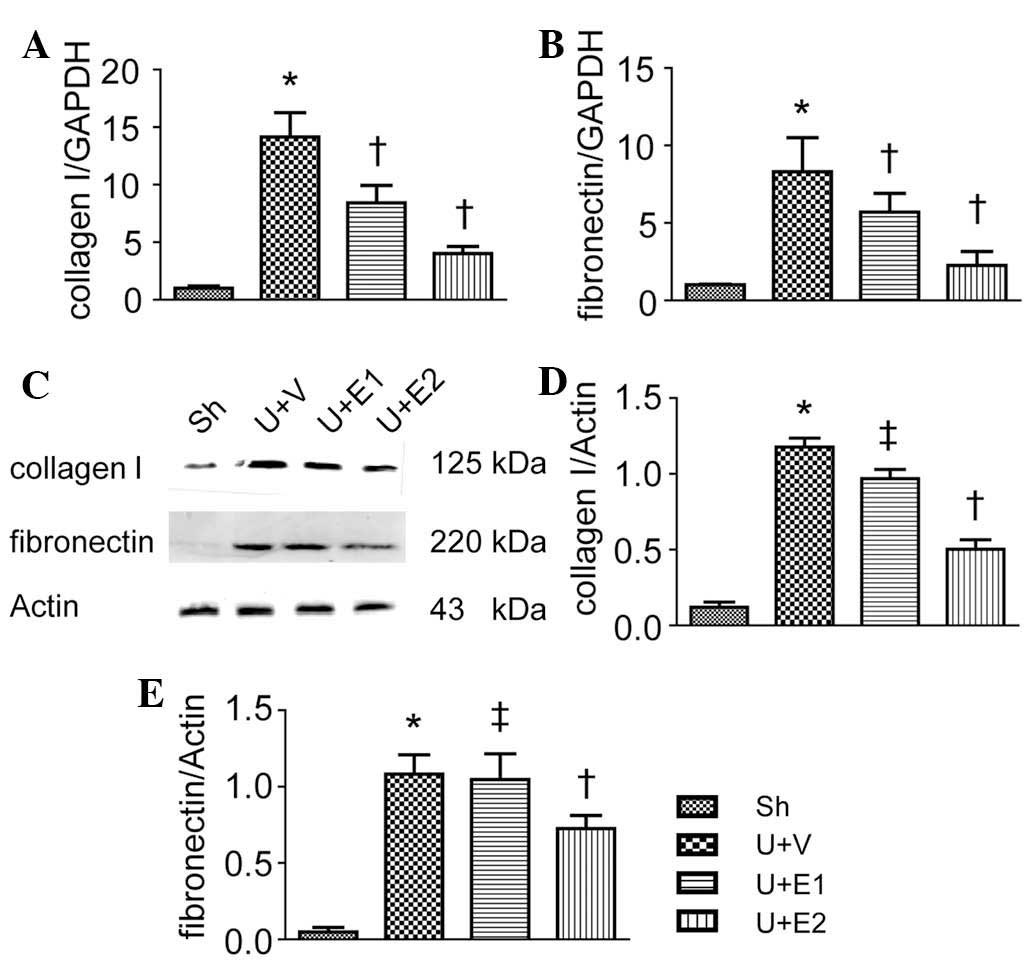
rhEPO-treatment suppressed renal collagen I and fibronectin
expression at the mRNA and protein levels (Fig. 2). These results indicated an
anti-fibrotic effect of rhEPO.
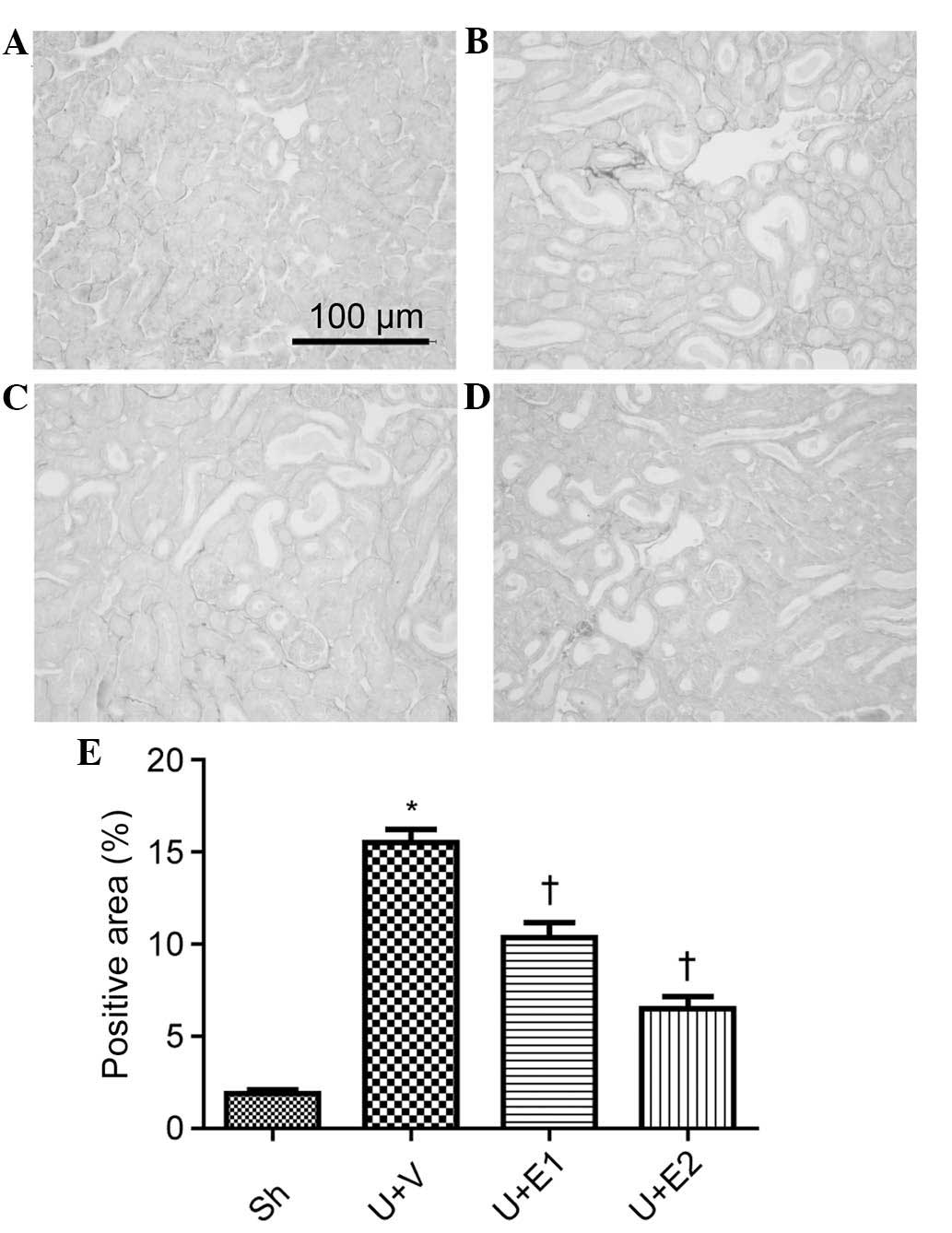 | Figure 1Renal interstitial fibrosis was
examined by picrio-sirus red staining on day seven post-surgery.
(A) Sh group, (B) U+V group, (C) U+E1 group, (D) U+E2 group. (E)
Semi-quantitative assessment of renal fibrosis. Values are
expressed and the mean ± standard deviation. *P<0.05
vs. the Sh group; †P<0.05 vs. U+V group. UUO,
unilateral ureteral obstruction; Sh, control; U+V, UUO+vehicle;
U+E1, UUO treated with 300 U/kg rhEPO; rhEPO, recombinant human
erythropoietin; U+E2, UUO treated with 1,000 U/kg rhEPO. |
EPO treatment attenuates myofibroblast
accumulation
Myofibroblasts are the effector cells for ECM
production; therefore, whether EPO treatment decreased
myofibroblast accumulation was evaluated. The control kidneys
revealed an absence of or weak labeling of α-SMA (Fig. 3A), which surrounded the major blood
vessels, whereas the vehicle-treated UUO kidneys demonstrated
increased expression of α-SMA in the interstitium (Fig. 3B). However, rhEPO-treatment (groups
U+E1 and U+E2) attentuated the accumulation of α-SMA positive cells
(Fig. 3C and D). Consistent with
the results of the immunohistochemical analysis, immunoblotting
revealed an increase in α-SMA expression levels in the obstructed
kidneys of the vehicle-treated UUO mice in comparison to those of
the control group. By contrast, rhEPO treatment significantly
inhibited α-SMA upregulation (Fig. 3E
and F).
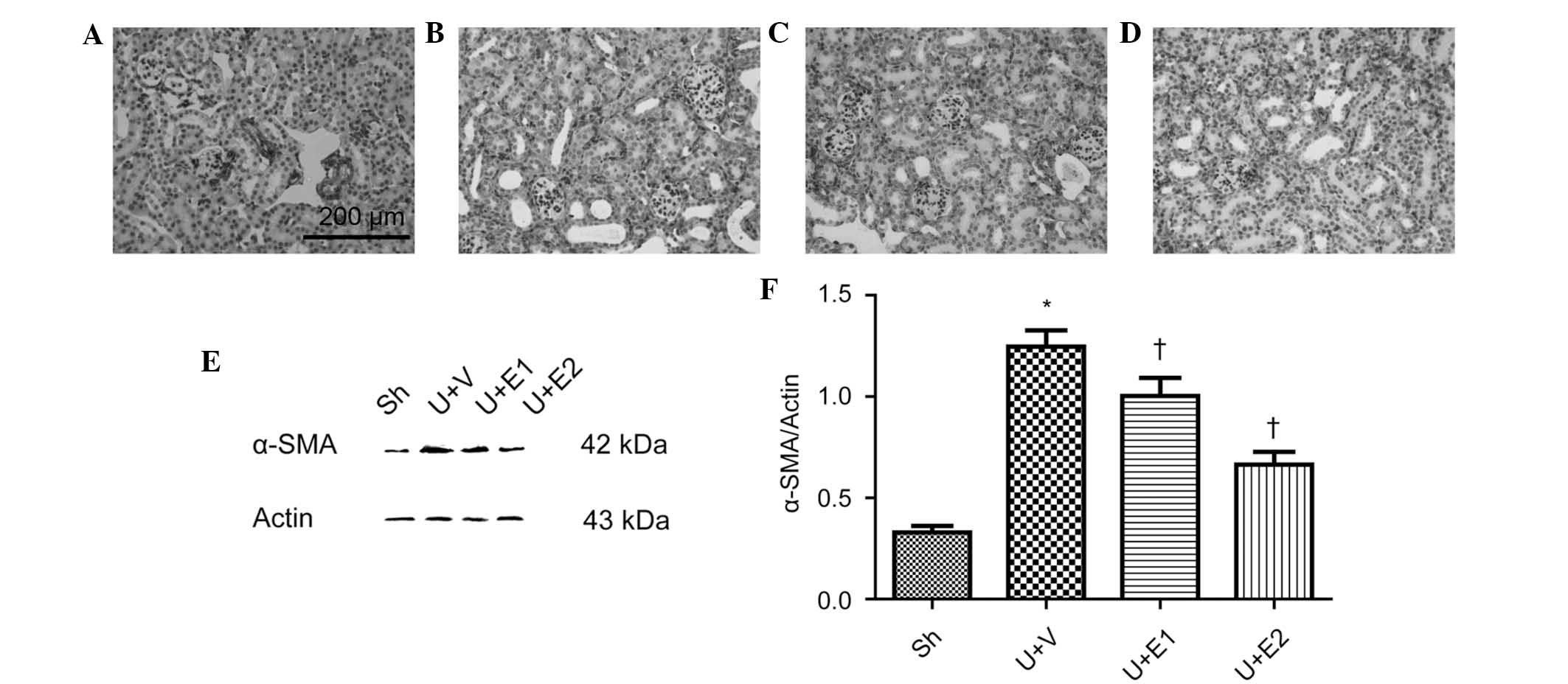 | Figure 3Representative photographs of α-SMA
immunohistochemistry on day 7 post-surgery. (A) Sh, (B) U+V, (C)
U+E1 and (D) U+E2 groups. (E) Representative gels of the western
blot analysis of α-SMA expression levels. (F) Semi-quantitative
analysis of α-SMA protein expression. Actin was used as an internal
control. *P<0.05 vs. Sh; †P<0.05 vs.
U+V. Values are expressed as the mean ± standard deviation. Scale
bar, 200 μm. UUO, unilateral ureteral obstruction; Sh, control;
U+V, UUO+vehicle; U+E1, UUO treated with 300 U/kg rhEPO; rhEPO,
recombinant human erythropoietin; U+E2, UUO treated with 1,000 U/kg
rhEPO; α-SMA, α-smooth muscle actin. |
EPO treatment decreases fibrocyte
accumulation in the interstitium
Myofibroblasts are potentially derived from multiple
sources, so whether rhEPO treatment was able to decrease fibrocyte
accumulation was investigated. Compared with the control group, UUO
increased fibrocyte accumulation in the interstitium, as detected
by flow cytometry (Fig. 4Aa, Ab and
B). By contrast, rhEPO (U+E1 and U+E2 groups) treatment
significantly inhibited the accumulation of fibrocytes (Fig. 4Ac, Ad and B).
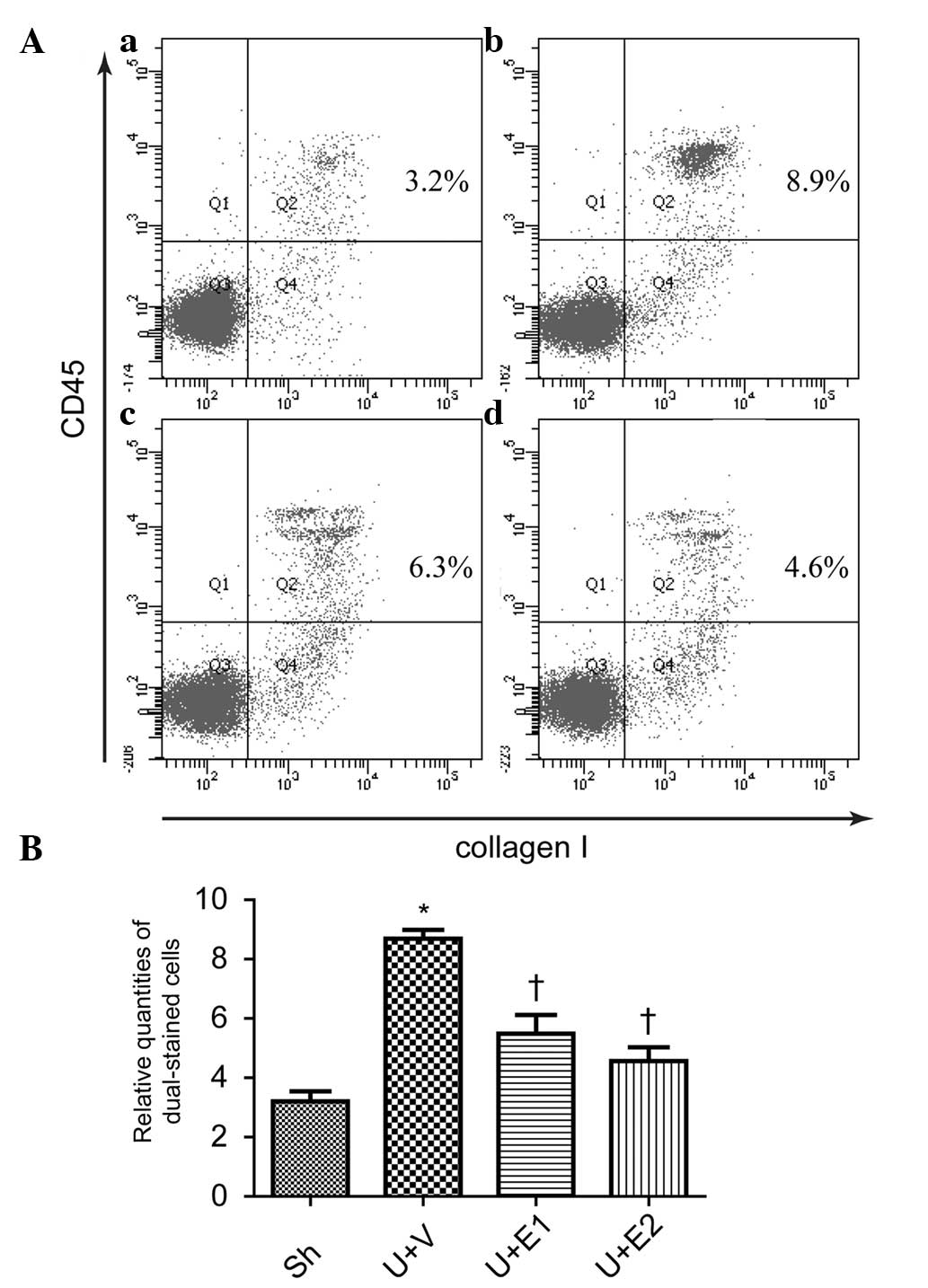 | Figure 4Fibrocytes in the interstitium were
detected by flow cytometry, indicated by the percentage of cells
positively dual-stained for collagen I and CD45. (A) Representative
photographs of (a) Sh, (b) U+V, (c) U+E1 and (d) U+E2. (B)
Semi-quantitative analysis of the percentage of dual-stained cells.
*P<0.05 vs. Sh; †P<0.05 vs. U+V. UUO,
unilateral ureteral obstruction; Sh, control; U+V, UUO+vehicle;
U+E1, UUO treated with 300 U/kg rhEPO; rhEPO, recombinant human
erythropoietin; U+E2, UUO treated with 1,000 U/kg rhEPO. |
EPO decreases CXCL16 expression
Bone marrow-derived fibrocytes migrate into the
kidney by following chemoattractants, including CXCL12, CCL21 and
CXCL16. Whether rhEPO treatment was able to alter the expression of
these chemokines was examined. Compared with the control group,
increases in the expression of CXCL12, CCL21 and CXCL16 were
observed following UUO. However, rhEPO treatment (U+E1 and U+E2
groups) decreased the expression of CXCL16 but did not influence
CXCL12 or CCL21 expression (Fig.
5).
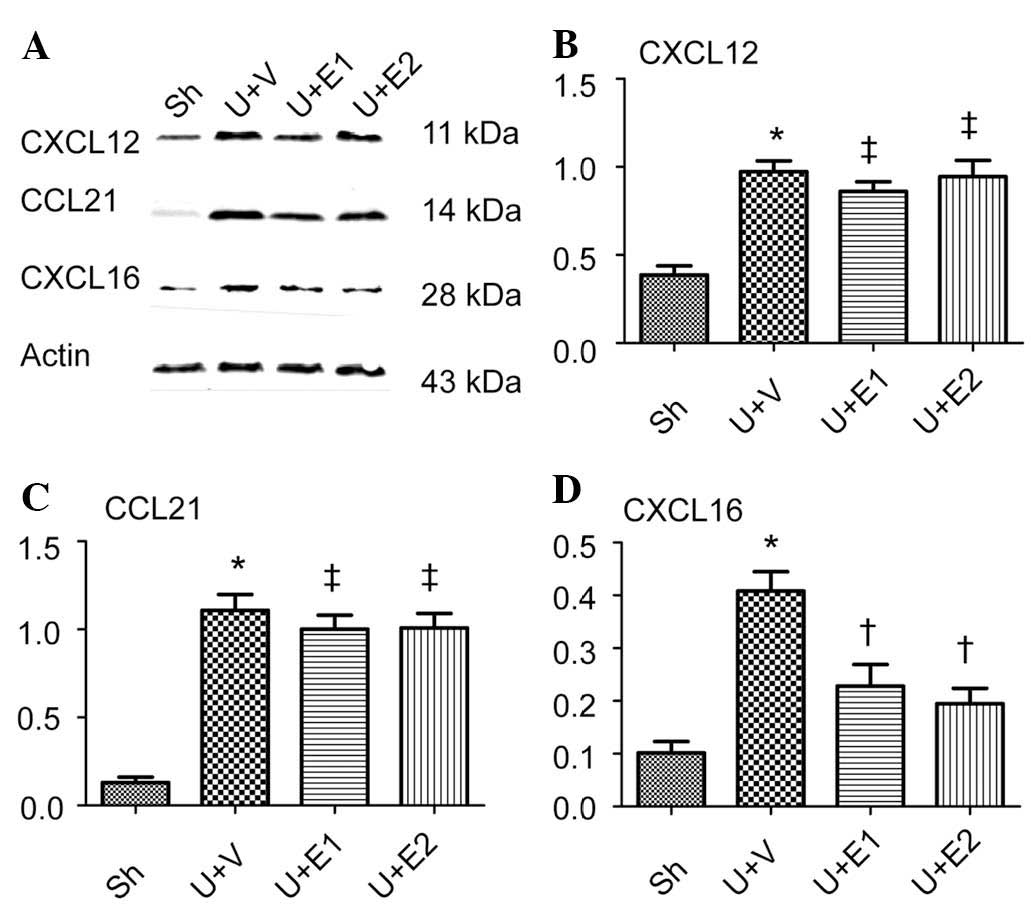 | Figure 5CXCL12, CCL21 and CXCL16 expression
analysis. (A) Representative western blots for CXCL12, CCL21 and
CXCL16. Semi-quantitative analysis of protein expression levels of
(B) CXCL12, (C) CCL21 and (D) CXCL16. Actin was used as an internal
control. *P<0.05 vs. Sh; †P<0.05 vs.
U+V and ‡P>0.05 vs. U+V. UUO, unilateral ureteral
obstruction; Sh, control; U+V, UUO+vehicle; U+E1, UUO treated with
300 U/kg rhEPO; rhEPO, recombinant human erythropoietin; U+E2, UUO
treated with 1,000 U/kg rhEPO; CXCL16, CXC chemokine ligand 16;
CCL21, CC chemokine ligand 21. |
Discussion
At present, no specific therapeutic modalities for
chronic renal disease are available (23). However, the prevention or
inhibition of renal fibrosis is one therapeutic option that may
potentially be capable of slowing, or even stopping the progression
of chronic renal disease. In the present study, it was demonstrated
that rhEPO ameliorated renal fibrosis in mice following complete
UUO. Furthermore, rhEPO decreased the expression of CXCL16 and the
accumulation of fibrocytes in the obstructed kidneys. Therefore, in
addition to its function in the treatment of anemia, the results of
the present study suggested that rhEPO treatment may be used in the
inhibition of renal fibrosis, in part via the attenuation of
fibrocyte accumulation in the kidneys.
In order to analyze the anti-fibrotic effects of
rhEPO, a UUO mouse model was used. The UUO mouse model is a well
established animal model for the induction of renal fibrosis and
assessment of the anti-fibrotic effects of various agents. The
model remains free of confounding factors, including exogenous
toxins or uremia, as the unobstructed kidney maintains normal
homeostasis and renal function (19). ECM was examined using PSR staining,
which indicated rhEPO decreased ECM accumulation. This result was
corroborated by PCR and western blot analyses of collagen I and
fibronectin expression. These results suggested an anti-fibrotic
effect of rhEPO.
The α-SMA positive cells are the main effectors
contributing to renal fibrosis (24). The results of the present study
demonstrated that rhEPO treatment attenuated the accumulation of
α-SMA positive cells, which may partially explain the anti-fibrotic
effects of rhEPO. However, the origin of the myofibroblasts
responsible for the excessive production of ECM has remained
elusive (25). A previous study
demonstrated that EMT may be an essential process in renal
fibrogenesis (26). Multiple
studies have provided evidence that bone marrow-derived
myofibroblasts may be recruited to the kidney and subsequently
contribute to kidney fibrosis (6–8,10,11,27,28).
Bone marrow-derived myofibroblast precursor cells,
named ‘fibrocytes’, were initially identified in the peripheral
circulation (29). These
fibrocytes were found to co-express mesenchymal markers, including
collagen I and vimentin, as well as hematopoietic markers,
including CD45, CD11b and CD34 (30). In culture, fibrocytes exhibited an
adherent, spindle-like morphology and expressed α-SMA following
treatment with TGFβ1, which was consistent with the hypothesis that
these cells are able to differentiate into myofibroblasts (4). In the present study, bone
marrow-derived myofibroblast precursors were demonstrated to
accumulate in the kidney following obstructive injury. Flow
cytometric analysis indicated that UUO induced CD45 and collagen I
dual-positive myofibroblast precursors to migrate into the injured
kidneys, an effect which was abrogated following rhEPO treatment.
These results suggested that the anti-fibrotic effect of rhEPO may
be partially due to the decreased accumulation of fibrocytes in the
obstructed kidneys.
The signaling mechanisms underlying the recruitment
of bone marrow-derived myofibroblast precursors into the kidney
remain to be elucidated (31).
Chemokines have significant roles in the regulation of
myofibroblast precursor infiltration in response to injury.
Chemokines are classified into four major families: C, CC, CXC and
CX3C, based on the relative position of cysteine residues proximal
to the amino terminus (31).
Chemokines activate seven-transmembrane G-protein-coupled receptors
and have primary functions in the trafficking of circulating cells
during inflammation.
Sakai et al (8) demonstrated that CCL21 and its
receptor, CCR7, were involved in the infiltration of myofibroblast
precursors into the kidney in a mouse model of renal fibrosis
induced by obstructive injury. A further study identified a role
for CXC12 in the accumulation of fibrocytes in animal models of
injury-induced pulmonary fibrosis (32). In addition, Chen et al
(9) revealed that the CXCL16-CXCR6
axis was associated with fibrocyte recruitment. The present study
aimed to evaluate whether chemokines CXCL12, CXCL16 and CCL21 were
engaged in fibrocyte recruitment. The results indicated that UUO
induced upregulation of CXCL12, CXCL16 and CCL21, and rhEPO
treament abrogated the upregulation of CXCL16, but did not
influence the expression of CXCL12 and CCL12. These results
suggested that rhEPO decreased renal CXCL16 expression, indicating
that this is may be one of the mechanisms underlying the reduction
of fibrocyte accumulation in the interstitium.
A low dosage of rhEPO decreased the mRNA expression
levels of collagen I and fibronectin, but did not alter the protein
expression levels. It was postulated that rhEPO influenced the
post-transcriptional processes. In addition, it was demonstrated
that a low dose of rhEPO ameliorated renal fibrosis, but did not
decrease the number of α-SMA-positive cells. This inconsistency may
be due to the fact that α-SMA positive cells are the main cells
responsible for ECM production, but are not the only cells involved
(33). The effects of long-term
rhEPO treatment on fibrocyte accumulation remain to be elucidated
and requires further investigation.
In conclusion, it was demonstrated that rhEPO
attenuated fibrocyte accumulation in the interstitium, indicating
that rhEPO may present a potential strategy for the prevention of
interstitial fibrosis and slow the progression of chronic renal
insufficiency. The model used in the present study indicated that
rhEPO decreased CXCL16, a chemokine that attracts circulating
fibroblast-like precursors, and attenuated the accumulation of bone
marrow-derived myofibroblasts. To the best of our knowledge, this
is the first study to report the beneficial effects of rhEPO
treatment, beyond hematopoiesis, against renal fibrosis via the
inhibition of fibrocyte accumulation. These results may therefore
provide novel insights into the mechanisms underlying the
protection conferred by treatment with rhEPO against chronic kidney
disease.
References
|
1
|
Duffield JS: Cellular and molecular
mechanisms in kidney fibrosis. J Clin Invest. 124:2299–2306. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Boor P and Floege J: The renal (myo-)
fibroblast: a heterogeneous group of cells. Nephrol Dial
Transplant. 27:3027–3036. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grgic I, Duffield JS and Humphreys BD: The
origin of interstitial myofibroblasts in chronic kidney disease.
Pediatr Nephrol. 27:183–193. 2012. View Article : Google Scholar
|
|
4
|
Bellini A and Mattoli S: The role of the
fibrocyte, a bone marrow-derived mesenchymal progenitor, in
reactive and reparative fibroses. Lab Invest. 87:858–870. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pilling D and Gomer RH: Differentiation of
circulating monocytes into fibroblast-like cells. Methods Mol Biol.
904:191–206. 2012.PubMed/NCBI
|
|
6
|
Iwano M, Plieth D, Danoff TM, Xue C, Okada
H and Neilson EG: Evidence that fibroblasts derive from epithelium
during tissue fibrosis. J Clin Invest. 110:341–350. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li J, Deane JA, Campanale NV, Bertram JF
and Ricardo SD: The contribution of bone marrow-derived cells to
the development of renal interstitial fibrosis. Stem Cells.
25:697–706. 2007. View Article : Google Scholar
|
|
8
|
Sakai N, Wada T, Yokoyama H, et al:
Secondary lymphoid tissue chemokine (SLC/CCL21)/CCR7 signaling
regulates fibrocytes in renal fibrosis. Proc Natl Acad Sci USA.
103:14098–14103. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen G, Lin SC, Chen J, et al: CXCL16
recruits bone marrow-derived fibroblast precursors in renal
fibrosis. J Am Soc Nephrol. 22:1876–1886. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Reich B, Schmidbauer K, Rodriguez Gomez M,
et al: Fibrocytes develop outside the kidney but contribute to
renal fibrosis in a mouse model. Kidney Int. 84:78–89. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang J, Lin SC, Chen G, et al: Adiponectin
promotes monocyte-to-fibroblast transition in renal fibrosis. J Am
Soc Nephrol. 24:1644–1659. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Oba S, Suzuki E, Nishimatsu H, et al:
Renoprotective effect of erythropoietin in ischemia/reperfusion
injury: possible roles of the Akt/endothelial nitric oxide
synthase-dependent pathway. Int J Urol. 19:248–255. 2012.
View Article : Google Scholar
|
|
13
|
Kaynar K, Aliyazioglu R, Ersoz S, et al:
Role of erythropoietin in prevention of amikacin-induced
nephropathy. J Nephrol. 25:744–749. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang W and Zhang J: Protective effect of
erythropoietin against aristolochic acid-induced apoptosis in renal
tubular epithelial cells. Eur J Pharmacol. 588:135–140. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Imamura R, Isaka Y, Sandoval RM, et al: A
nonerythropoietic derivative of erythropoietin inhibits
tubulointerstitial fibrosis in remnant kidney. Clin Exp Nephrol.
16:852–862. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen CL, Chou KJ, Lee PT, et al:
Erythropoietin suppresses epithelial to mesenchymal transition and
intercepts Smad signal transduction through a MEK-dependent
mechanism in pig kidney (LLC-PK1) cell lines. Exp Cell Res.
316:1109–1118. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kitamura H, Isaka Y, Takabatake Y, et al:
Nonerythropoietic derivative of erythropoietin protects against
tubulointerstitial injury in a unilateral ureteral obstruction
model. Nephrol Dial Transplant. 23:1521–1528. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Park SH, Choi MJ, Song IK, et al:
Erythropoietin decreases renal fibrosis in mice with ureteral
obstruction: role of inhibiting TGF-beta-induced
epithelial-to-mesenchymal transition. J Am Soc Nephrol.
18:1497–1507. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chevalier RL, Forbes MS and Thornhill BA:
Ureteral obstruction as a model of renal interstitial fibrosis and
obstructive nephropathy. Kidney Int. 75:1145–1152. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Grande MT, Fuentes-Calvo I, Arévalo M, et
al: Deletion of H-Ras decreases renal fibrosis and myofibroblast
activation following ureteral obstruction in mice. Kidney Int.
77:509–518. 2010. View Article : Google Scholar
|
|
21
|
Kim DH, Moon SO, Jung YJ, et al: Mast
cells decrease renal fibrosis in unilateral ureteral obstruction.
Kidney Int. 75:1031–1038. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li L, Huang L, Sung SS, et al: The
chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage
trafficking in kidney ischemia-reperfusion injury. Kidney Int.
74:1526–1537. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Formentini I, Bobadilla M, Haefliger C, et
al: Current drug development challenges in chronic kidney disease
(CKD) - identification of individualized determinants of renal
progression and premature cardiovascular disease (CVD). Nephrol
Dial Transplant. 27(Suppl 3): iii81–iii88. 2012. View Article : Google Scholar
|
|
24
|
Strutz F and Zeisberg M: Renal fibroblasts
and myofibroblasts in chronic kidney disease. J Am Soc Nephrol.
17:2992–2998. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Meran S and Steadman R: Fibroblasts and
myofibroblasts in renal fibrosis. Int J Exp Pathol. 92:158–167.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kriz W, Kaissling B and Le Hir M:
Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or
fantasy? J Clin Invest. 121:468–474. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Broekema M, Harmsen MC, van Luyn MJ, et
al: Bone marrow-derived myofibroblasts contribute to the renal
interstitial myofibroblast population and produce procollagen I
after ischemia/reperfusion in rats. J Am Soc Nephrol. 18:165–175.
2007. View Article : Google Scholar
|
|
28
|
Niedermeier M, Reich B, Rodriguez Gomez M,
et al: CD4+ T cells control the differentiation of Gr1+ monocytes
into fibrocytes. Proc Natl Acad Sci USA. 106:17892–17897. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bucala R, Spiegel LA, Chesney J, Hogan M
and Cerami A: Circulating fibrocytes define a new leukocyte
subpopulation that mediates tissue repair. Mol Med. 1:71–81.
1994.PubMed/NCBI
|
|
30
|
Wada T, Sakai N, Matsushima K and Kaneko
S: Fibrocytes: a new insight into kidney fibrosis. Kidney Int.
72:269–273. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chung AC and Lan HY: Chemokines in renal
injury. J Am Soc Nephrol. 22:802–809. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Phillips RJ, Burdick MD, Hong K, et al:
Circulating fibrocytes traffic to the lungs in response to CXCL12
and mediate fibrosis. J Clin Invest. 114:438–446. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Boor P, Ostendorf T and Floege J: Renal
fibrosis: novel insights into mechanisms and therapeutic targets.
Nat Rev Nephrol. 6:643–656. 2010. View Article : Google Scholar : PubMed/NCBI
|