Introduction
The hepatitis C virus (HCV) is a member of the
Flaviviridae family of positive-strand RNA viruses and encodes 10
proteins, including three structural proteins (the core protein and
the envelope glycoproteins E1 and E2) and seven nonstructural (NS)
proteins (NS2, NS3, NS4A, NS4B, NS5A and NS5B) (1–3). The
NS2 protein, derived from the cleavage of NS2/3, is inserted into
the endoplasmic reticulum membrane through its N-terminal
hydrophobic domain (4). NS2 is a
hydrophobic protein containing several transmembrane segments in
the N-terminal region. NS2 may also be involved in modulating
cellular gene expression in infected cells (5,6),
although the molecular mechanisms remain to be elucidated.
Suppression subtractive hybridization (SSH) has been
previously used to successfully identify and isolate differentially
expressed genes (7), particularly
the isolation of rare transcripts. To assign a role for NS2, the
present study aimed to identify cellular proteins, which interacted
with NS2. Coiled-coil-heli x-coiled-coil-helix domain-containing
protein 2 (CHCHD2) is a member of a protein family containing a
(coiled-coil 1)-(heli x 1)-(coiled-coil 2)-(helix 2) (CHCH) domain
(8,9). Our group revealed that the protein
was expressed in HCV (10);
however, to the best of our knowledge, its expression in liver
cancer had not been identified. The present study therefore aimed
to investigate CHCHD2 expression in liver cancer. The first and
second coiled-coil regions of the CHCH domain have a fixed length
of 10 amino acid (aa) and a variable length of 5–10 aa,
respectively. The second coiled-coil region may act as a bridge
when the two helices fold towards each other. Each α-helix within
the CHCH domain contains two cysteine aas (11), which are separated either by nine
residues (CX9C motif) (11)
observed in the mitochondrial intermembrane space protein Mia40,
cytochrome c oxidase copper chaperone, cyclooxygenase 1 and
cytochrome c oxidase subunit VIIa or by three residues (CX3C motif)
observed in the translocase of inner membrane proteins. Two
interhelica l disulfide bonds may contribute to the formation and
stabilization the protein tertiary structure (9).
The transcription factor cyclic adenosine
monophosphate (cAMP) response element-binding protein (CREB) is a
member of a leucine zipper class of transcription factors, which
specifically recognizes the cAMP-response element (CRE) promoter
site (12). CREB is a target of
other signaling pathways and is activated by a diverse array of
stimuli, including peptide hormones, growth factors and neuronal
activity, which activate a variety of protein kinases, including
protein kinase A (PKA), PKC, mitogen-activated protein kinases
(MAPKs) and Ca2+/calmodulin-dependent protein kinases
(13,14). Previous studies have indicated that
the phosphorylation of Ser133 is required for CREB-induced gene
transcription. Phosphorylation at Ser133 may induce the
translocation of cytoplasmic CREB to the nucleus. Phosphorylated
CREB within the nucleus may lead to transcriptional activation by
promoting interaction with components of the basal transcription
machinery, including transcription factor II D and RNA polymerase
II (15,16). In addition, there are residues in
addition to Ser-133 and the post-translational modifications of
CREB (17). CREB regulates several
cellular functions, including inflammation, cell proliferation,
differentiation, adaptation and survival. However, the role of CREB
in regulating the expression of CHCHD2 remains to be
elucidated.
The function of CHCHD2 is complex and it is likely
that the control of CHCHD2 occurs at multiple levels. However, the
mechanisms regulating the expression of CHCHD2 remain to be
elucidated.
Materials and methods
Clinical specimens
Patients with a diagnosis of HCC underwent
hepatectomy surgery between January 2009 and May 2012. In total,
110 samples were obtained from these surgeries. The mean patient
age was 50.9±9.2 years, 87 patients were male and 23 were female.
All were obtained from paraffin blocks from Beijing Ditan Hospital.
HCC paraffin blocks and frozen tissues were obtained from the
archives of the Department of Pathology, Beijing Ditan Hospital,
Capital Medical University (Beijing, China) with approval from the
Institutional Review Board. A total of 110 HCC tumor and 50
noncancerous liver tissue samples, which included 20 patients with
cirrhosis, 20 patients with other liver diseases, including chronic
hepatitis, drug-induced liver injury and nonalcoholic
steatohepatitis and 10 healthy controls were obtained.
Clinical follow-up data, including serum levels of
α-fetoprotein (AFP) and other blood biochemical indices were
obtained from the patients’ records and retrospectively from case
review.
The use of human specimens in the present study was
approved by the Ethics Committee of Beijing Ditan Hospital
according to the Declaration of Helsinki. All necessary consent was
obtained from any patients involved in the present study, including
consent for involvement in the study and consent to publish.
Immunohistochemistry
The tissue specimens were routinely processed and
stained with hematoxylin and eosin (H&E). The diagnosis of HCC
was based on pathology according to the International Working Party
criteria (18) and tumor grading
was assessed on the H&E-stained sections according to Scheuer’s
system (18).
The HCC biopsy specimens were subjected to routine
immunohistochemical staining using a monoclonal antibody according
to a previously described method (19). Briefly, sections were
deparaffinized in xylene and rehydrated in decreasing
concentrations of ethanol (100, 90, 80 and 70%). Slides were
incubated in 3% H2O2, 50% methanol in wash
buffer [phosphate-buffered saline (PBS) and 0.1% Tween-20] to
quench endogenous peroxidases. Sections were subsequently incubated
with primary antibodies for 1h at room temperature. Primary
antibodies were purchased from Abcam (Cambridge, UK), and included
monoclonal mouse anti-human CD34 (1:50), GPC3 (1:10), GS (1:100),
HSP70 (1:100), Ki-67 (1:75) and CHCHD2 (1:300), and the PEG10
(1:1,000) anti-mouse secondary antibody. All antibodies were
obtained from Dako North America, Inc. (Carpinteria, CA, USA).
Immunoreactivity, which was defined as the number of
positive tumor cells observed in the total number of tumor cells,
was scored independently by two researchers. The numbers of
CHCHD2-positive and -negative HCC cells were counted under a light
microscope (80i; Olympus Corp., Tokyo, Japan) at a magnification of
×400, with only the cells exhibiting brown nucleoli considered
CHCHD2-positive. For each slide, between 7 and 10 microscopic
fields were randomly selected. The positive scores were the
categorized into weak staining with staining in only one nucleolus;
moderate staining, with staining in more than one nucleolus; and
strong staining, with staining present in the nucleus and nucleolus
of the tumor cell. The average percentage of CHCHD2-positive HCC
cells was then calculated for each group.
Cell culture
The HepG2 cells (China Infrastructure of Cell Line
Resource, Beijing, China) were cultured in Dulbecco’s modified
Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum
(Shanghai BioAsia Biotechnology, Shanghai, China), 100 U/ml
penicillin G and 100 μg/ml streptomycin (North China
Pharmaceutical Co. Ltd, Shijiazhuang, China) in a humidified
chamber at 37°C and 5% CO2. The cells were seeded into
48-well plates, grown to 90% confluence and transiently transfected
using Lipofectamine 2000 (Invitrogen Life Technologies, Carlsbad,
CA, USA) according to the manufacturer’s instructions.
Screening of SSH, clones and
identification of the CHCHD2 gene
A subtracted cDNA library was constructed by SSH
using a PCR-select™ cDNA subtraction kit (Clontech Laboratories,
Inc., Mountain View, CA, USA) according to the manufacturer’s
instructions. A total of 2 μg mRNA was used for each
first-strand cDNA synthesis. The cDNA obtained from the HepG2 cells
transfected with pcDNA3.1 (−)−NS2 (genotype 1b; Beijing Key
Laboratory of Emerging Infectious Diseases, Beijing Ditan Hospital)
was used as a tester and the cDNA from the HepG2 cells transfected
with pcDNA3.1 (−) was used as a driver. Subsequently, the
subtracted PCR products were cloned into pGEM-T Easy vectors
(Promega Corp., Madison, WI, USA). The ligation reactions were then
transformed into chemically competent DH5α cells (China
Infrastructure of Cell Line Resource) using standard molecular
biology techniques.
Following sequencing of the positive colonies
(Shanghai BioAsia Biotechnology, Shanghai, China), the basic local
alignment search tool (BLAST) server (http://blast.ncbi.nlm.nih.gov/Blast.cgi) at the
National Center for Biotechnology Information (Bethesda, MD, USA)
was used to identify nucleic acid homology.
Expression of CHCHD2
The HepG2 cells were lysed for 36 h by transient
transfection with Mammalian Cell Lysis reagent (Thermo Fisher
Scientific, Waltham, MA, USA) containing 1% phenylmethyl
sulfonylfluoride and centrifuged for 5 min at 12,000 × g. The
lysates were then diluted in the supernatant using sodium dodecyl
sulfate (SDS) buffer and separated using SDS-PAGE (Thermo Fisher
Scientific). Following transfer onto a nitrocellulose membrane
(Millipore, Billerica, MA, USA), the blots were inhibited for 2 h
with 5% milk-PBST (PBS+0.05% Tween). For the detection of CHCHD2,
rabbit CHCHD2 antibody (1.0 mg/ml) (20) was diluted (1:1,000) in PBST buffer.
The mouse anti-β-actin antibody (Santa Cruz Biotechnology, Inc.,
Austin, TX, USA) was diluted (1:2,000) in PBST buffer. After 10 h
incubation at 4°C and extensive washing with PBST buffer,
horseradish peroxidase-conjugated goat-anti-rabbit immunoglobulin
(Ig) secondary antibody (Bio-Rad, Hercules, CA, USA) was added
(1:1,000) in 5% milk-PBST buffer for 2 h at room temperature, which
was also used to detect the protein expression of β-actin.
Following additional washing, the blots were developed using an
enhanced chemiluminescence substrate system (Pierce ECL Western
Blotting Substrate; Thermo Fisher Scientific).
RNA extraction and reverse transcription
quantitative polymerase chain reaction (RT-qPCR) analysis
The total RNA was extracted from the cultured cells
using TRIzol reagent (Promega Corp.) according to the
manufacturer’s instructions. A total of 0.1 μg RNA from each
sample was used to generate cDNA by reverse transcription using the
One-step RT-PCR kit (Takara Bio, Inc., Shiga, Japan). A Taqman
RT-qPCR assay was performed on an ABI Prism 7500 system (Applied
Biosystems Life Technologies, Foster City, CA, USA) according to
the manufacturer’s instructions. β-actin was used as a reference
for normalizing the data. The cycling conditions were as follows:
95°C for 30 sec, 40 cycles of 95°C for 5 sec, 60°C for 34 sec and
finally 60°C for 1 min. The CHCHD2 primers and probe used were as
follows: Forward, 5′-GCATCATCCCCATTCCGAAGG-3′ and reverse,
5′-ACCTGATTGGCTCGCTCTCC-3′; probe: 5′-CTCCGGCTGCACCTCGCTTGGC-3′.
The GAPDH primers used were: Forward, 5′-ACAGCCTCAAGATCA TCAGCA-3′
and reverse, 5′-ATGAGTCCTTCCACGATA CCA-3′; probe:
5′-GTGCTAAGCAGTTGGTGGTGCAGG A-3′. Primers were obtained from
Promega Corp.
Molecular cloning of the CHCHD2
promoter
The genomic DNA was isolated from the HepG2 cells
using a Genomic DNA Purification kit (Promega Corp.). A series of
5′-fanking DNA fragments upstream of the transcription initiation
site of CHCHD2, N1 (between −1871 and +93), N2 (between −1691 and
+93), N3 (between −257 and +93) and N4 (between −157 and +93) were
inserted into the Kpn I and Bgl II restriction sites of a pGL4.10
Basic vector (Promega Corp.). The PCR primers used were as follows:
N1 forward, 5′-GG TACCCTTTGGGGGGAACAGGTGGT-3′; N2 forward,
5′-GGTACCACCCACCTAGCACATCCC-3′; N3 forward,
5′-GGTACCGTTGACCGCGAAGGACGAG-3′ and N4 forward,
5′-GGTACCTGGTTGGTTGCGCGTTGAG-3′; as well as a common reverse,
5′-AGATCTCGGCCTCCC TCTGCGTCAT-3′.
The coding sequence of CREB was amplified from the
HepG2 cDNA by qPCR and was subcloned into pcDNA3.1 (−). The primers
used were as follows: Forward, 5′-GAATTCCGGAGGTGTAGTTTGACG-3′ and
reverse, 5′-GGATCCTTAATCTGATTTGTGGCAGT-3′.
Transient transfection and luciferase
reporter assays
The HepG2 cells were cotransfected with 0.4
μg plasmid-constructed CHCHD2 promoter and 13 ng internal
control plasmid phRL-TK using Lipofectamine 2000 (Invitrogen Life
Technologies) according to the manufacturer’s instructions. At 24 h
post-transfection, the cells were harvested and lysed in 50
μl passive lysis buffer (Thermo Fisher Scientific). A
fraction of the protein was subjected to a Dual-luciferase Reporter
Assay System kit (Promega Corp.). The firefly luciferase activity
and Renilla luciferase activity were measured sequentially using a
Veritas Microplate Luminometer (Turner BioSystems, Inc., Sunnyvale,
CA, USA). All transfections were performed in triplicate and the
promoter activities were expressed as the mean ± standard deviation
of three independent experiments.
Electrophoretic mobility shift assay
(EMSA)
For the gel shift assay, double-stranded DNA
oligonucleotides were synthesized with a biotin label at the 3′-end
(Invitrogen, Shanghai, China). The nuclear extracts were prepared
using a Nuclear Extraction kit (Pierce Biotechnology, Inc.,
Rockford, IL, USA) and the protein content was measured using a BCA
Protein Assay kit (Pierce Biotechnology, Inc.) according to the
manufacturer’s instructions. EMSAs were performed using a
LightShift chemiluminescence EMSA kit (Pierce Biotechnology, Inc.).
The oligonucleotide selected for EMSA contained sequences matching
a consensus CREB binding site. CREB wild-type oligonucleotide
probe, 5′-GGAAGAGCAGGACGTCACGGG GACGCCTCGTCC-3′ and mutant
oligonucleotide probe, 5′-GGAAGAGCAGGACCGGGGACGCCTCGTCC-3′. The
nuclear extracts, containing 5 μg protein, were incubated
with the aforementioned oligonucleotide probes for 20 min at 25°C.
For the super shift assay, 5 μg anti-CREB (Abcam, Cambridge,
MA, USA) was pre-incubated with the nuclear extracts for 30 min and
the labeled probes were then added to the reaction. Subsequently,
the DNA-protein complexes were separated using a 6.5%
non-denaturing polyacrylamide gel (Life Technologies, Grand Island,
NY, USA).
Chromatin immunoprecipitation (ChIP)
A total of 1×106 HepG2 cells were used
for each ChIP assay. The chromatin isolation and ChIP assays were
performed using an EZ-Zyme Chromatin prep kit and an EZ-ChIP kit
(Millipore). The chromatin solution was immunoprecipitated with
either 5 μg anti-CREB (Abcam) or 5 μg normal
anti-immunoglobulin (Ig)G antibody and 20 μl protein A
agarose beads (Cell Signaling Technology, Inc., Danvers, MA, USA)
overnight at 4°C. Following sequential washes, once with each of
the following buffers (Buffer A, low salt wash buffer; Buffer B,
high salt wash buffer; Buffer C, LiCl wash buffer and Buffer D,
Tris-HCl EDTA buffer), the antibody-protein-DNA complex was eluted
from the beads. Following reverse cross-link incubation, which
comprised the addition of 20 μl 5 M NaCl per tube (NaCl
final concentration, 0.2 M). The tubes were agitated continuously
and incubated at 65°C overnight to induce cross-linking.
Subsequently, the protein and RNA were removed by proteinase K and
RNase and a qPCR assay was performed on the immunoprecipitated
genomic DNA with primers specific for the CREB binding site
upstream of the transcriptional start site. The primers used were
as follows: Forward, 5′-AGGACCGGAGGACAAGGTTC-3′ and reverse,
5′-CTTCCGTTCTCCGTCGTCTC-3′.
Site-directed mutagenesis
N3 was used to perform site-directed mutagenesis of
the putative CREB binding sites following the quick change
site-directed mutagenesis protocol (Stratagene, La Jolla, CA, USA).
The oligonucleotide incorporating mutant bases was as follows:
CREB, 5′-GGACGAGGCGTCCCCGGTCCTGCTCTTCC-3′. The qPCR reaction was
performed for 30 cycles (95°C for 30 sec, 55°C for 30 sec and 68°C
for 10 min) following an initial denaturation at 95°C for 30 sec.
The mixture of input and amplified DNA was digested directly with
Dpn I and then transfected into the HepG2 cells. The nucleotide
sequences (5′-GCGTGATCCCTGGTACCAGAGCTCCGCCTC-3′) of the mutant were
confirmed by sequencing (Beijing AuGCT DNA-SYN Biotechnology Co.,
Ltd., Beijing, China).
Statistical analysis
All of the experiments were performed at least three
times. The results are expressed as the mean ± standard deviation.
Statistical comparisons were made using an unpaired two-tailed
Student’s t-test. P<0.05 was considered to indicate a
statistically significant difference. All analyses were performed
using SAS statisical software version 9.1 (SAS, Cary, NC, USA).
Results
Patients and clinical
characteristics
Table I shows the
characteristics of the 110 patients. Of these, the median age was
50.9±9.2 years and the majority of patients were male. As expected,
hepatitis B virus (HBV) was the major etiology (n=105) observed in
95.5% of the total patients. The majority of patients (n=96, 87.3%)
had evidence of cirrhosis. In total, >61 patients (55%) had not
received antiviral therapy; however, the mean duration of HBV was
12.1±8.6 years. Vascular invasion was relatively infrequent. The
AFP levels in the majority of patients with HCC was <20
μg/l.
 | Table ITumor and patient
characteristics. |
Table I
Tumor and patient
characteristics.
| A, Patient
characteristic | Number (%) |
|---|
| Age (years) | |
| Mean | 50.9±9.2 |
| Range | 30–80 |
| 30–40 | 15 (13.6) |
| 41–50 | 39 (35.4) |
| 51–60 | 43 (39.1) |
| >60 | 13 (11.8) |
| Gender | |
| Male | 87 (79.1) |
| Female | 23 (20.9) |
| Viral etiology | |
| Hepatitis B | 105 (95.5) |
| Hepatitis C | 5 (4.5) |
| Virus found
time | |
| Mean (years) | 12.1±8.6 |
| Longest | 40 years |
| Shortest | 1 day |
| Antiviral time
(years) | |
| ≤1 | 16 (14.5) |
| 1–3 | 10 (9.1) |
| >3 | 23 (20.9) |
| No antivirus | 61 (55.5) |
| Outcome (3
years) | |
| Alive | 77 (70.0) |
| Deceased | 7 (6.4) |
| Unknown | 26 (23.6) |
| HBV DNA level
(cp/ml) | |
| <500 | 51 (46.4) |
| ≥500 | 54 (49.1) |
|
| B, Tumor
characteristics |
|
| Diameter of tumor
(cm) | |
| <3 | 48 (43.6) |
| 3–5 | 45 (40.9) |
| >5 | 17 (15.5) |
| Cirrhosis | |
| Yes | 96 (87.3) |
| No | 14 (12.7) |
| AFP(ng/ml) | |
| ≤20 | 47 (42.7) |
| 20<AFP≤200 | 26 (23.6) |
|
200<AFP≤400 | 20 (18.2) |
| >400 | 17 (15.5) |
| Envelope | |
| Complete | 49 (44.5) |
| No envelope | 27 (24.5) |
| Unknown | 34 (30.9) |
| Lesion
position | |
| Right lobe | 83 (75.5) |
| Tumor
characteristic | Number (%) |
| Left lobe | 26 (23.6) |
| Caudate lobe | 1 (0.9) |
| Vascular
invasion | |
| Positive | 46 (41.8) |
| Negative | 64 (58.2) |
| Differentiation of
tumors | |
| High grade | 11 (10.0) |
| Moderate
grade | 63 (57.3) |
| Low grade | 36 (32.7) |
| Hepatitis B | 105 |
| HBeAg(+) | 46 (41.8) |
| HBeAg(-) | 59 (53.6) |
Molecular diagnosis of HCC
The results of the staining for gene expression
(CD34, GPC-3, GS, HSP70, Ki-67, PEG10 and CHCHD2) are shown in
Table II. The number of specimens
was limited; therefore, certain biomarkers were not determined in
all samples as single staining was performed on one slide. As
Table II shows, the CD34, GPC-3,
GS, HSP70, Ki-67 and PEG10 biomarkers had a positive rate of
>90% and the novel biomarker CHCHD2 was expressed in HCC. The
positive ratio for the presence of CHCHD2 was 96.3%; however, no
significant difference was observed compared with that of the other
biomarkers (P>0.05). All the biomarkers exhibited highly
positive rates for moderate- and low-grade HCC, whereas only CD34,
CHCHD2 and PEG10 exhibited high rates for high-grade HCC. The
expression levels of different biomarkers were associated with
tumor size, differentiation and AFP level; however, no significant
difference was observed between their expression levels (Table III).
| Table IIExpression of gene markers associated
with differentiation. |
Table II
Expression of gene markers associated
with differentiation.
| Gene marker | Positive, n (%)
| Negative, n (%)
|
|---|
| Total | Differentiation
| Total | Differentiation
|
|---|
| High | Moderate | Low | High | Moderate | Low |
|---|
| CD34 | 77 (100) | 6 (100) | 45 (100) | 26 (100) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| GPC-3 | 75 (94.9) | 5 (83.3) | 45 (95.7) | 25 (96.2) | 4 (5.1) | 1 (16.7) | 2 (4.3) | 1 (3.8) |
| GS | 37 (94.9) | 4 (80.0) | 19 (100) | 14 (93.3) | 2 (5.1) | 1 (20.0) | 0 (0.0) | 1 (6.7) |
| HSP70 | 38 (97.4) | 4 (80.0) | 19 (100) | 15 (100) | 1 (2.6) | 1 (20.0) | 0 (0.0) | 0 (0.0) |
| Ki-67 | 76 (96.2) | 5 (83.3) | 46 (97.8) | 25 (96.2) | 3 (3.8) | 1 (16.7) | 1 (2.1) | 1 (3.8) |
| CHCHD2 | 105 (96.3) | 10 (90.9) | 60 (95.2) | 35 (100) | 4 (3.7) | 1 (9.1) | 3 (4.8) | 0 (0.0) |
| PEG10 | 109 (99.1) | 11 (100) | 62 (98.4) | 36 (100) | 1 (0.9) | 0 (0.0) | 1 (1.6) | 0 (0.0) |
| Table IIIExpression of different gene markers
associated with tumor size, differentiation and AFP level. |
Table III
Expression of different gene markers
associated with tumor size, differentiation and AFP level.
| Size (cm) | Differentiation
| AFP
|
|---|
| High | Moderate | Low | <200 | 200–400 | >400 |
|---|
|
|
|---|
| + | – | + | – | + | – | + | – | + | – | + | – |
|---|
| CD34 | <3 | 4 | 0 | 23 | 0 | 10 | 0 | 25 | 0 | 5 | 0 | 7 | 0 |
| 3–5 | 2 | 0 | 14 | 0 | 11 | 0 | 20 | 0 | 4 | 0 | 3 | 0 |
| >5 | 0 | 0 | 8 | 0 | 5 | 0 | 6 | 0 | 2 | 0 | 5 | 0 |
| GPC-3 | <3 | 4 | 0 | 23 | 1 | 91 | 1 | 24 | 2 | 5 | 0 | 7 | 0 |
| 3–5 | 1 | 1 | 13 | 1 | 11 | 0 | 18 | 2 | 5 | 0 | 2 | 0 |
| >5 | 0 | 0 | 9 | 0 | 5 | 0 | 6 | 0 | 3 | 0 | 5 | 0 |
| GS | <3 | 3 | 0 | 10 | 0 | 5 | 0 | 13 | 0 | 0 | 0 | 5 | 0 |
| 3–5 | 1 | 1 | 5 | 0 | 7 | 1 | 8 | 2 | 3 | 0 | 2 | 0 |
| >5 | 0 | 0 | 4 | 0 | 2 | 0 | 1 | 0 | 0 | 0 | 5 | 0 |
| HSP70 | <3 | 3 | 0 | 10 | 0 | 5 | 0 | 13 | 0 | 0 | 0 | 5 | 0 |
| 3–5 | 1 | 1 | 5 | 0 | 8 | 0 | 9 | 1 | 3 | 0 | 1 | 0 |
| >5 | 0 | 0 | 4 | 0 | 2 | 0 | 1 | 0 | 0 | 0 | 5 | 0 |
| Ki-67 | <3 | 4 | 0 | 23 | 1 | 10 | 0 | 25 | 1 | 5 | 0 | 7 | 0 |
| 3–5 | 1 | 1 | 14 | 0 | 11 | 0 | 19 | 1 | 5 | 0 | 2 | 0 |
| >5 | 0 | 0 | 9 | 0 | 1 | 1 | 5 | 1 | 3 | 0 | 5 | 0 |
| CHCHD2 | <3 | 5 | 0 | 32 | 0 | 1 | 0 | 34 | 0 | 7 | 0 | 7 | 0 |
| 3–5 | 5 | 1 | 17 | 3 | 18 | 0 | 27 | 4 | 10 | 0 | 4 | 0 |
| >5 | 0 | 0 | 11 | 0 | 6 | 0 | 8 | 0 | 3 | 0 | 6 | 0 |
| PEG10 | <3 | 5 | 0 | 32 | 0 | 11 | 0 | 34 | 0 | 7 | 0 | 30 | 1 |
| 3–5 | 6 | 0 | 19 | 1 | 19 | 0 | 30 | 1 | 10 | 0 | 4 | 0 |
| >5 | 0 | 0 | 11 | 0 | 6 | 0 | 8 | 0 | 3 | 0 | 6 | 0 |
Expression of biomarkers in HCC
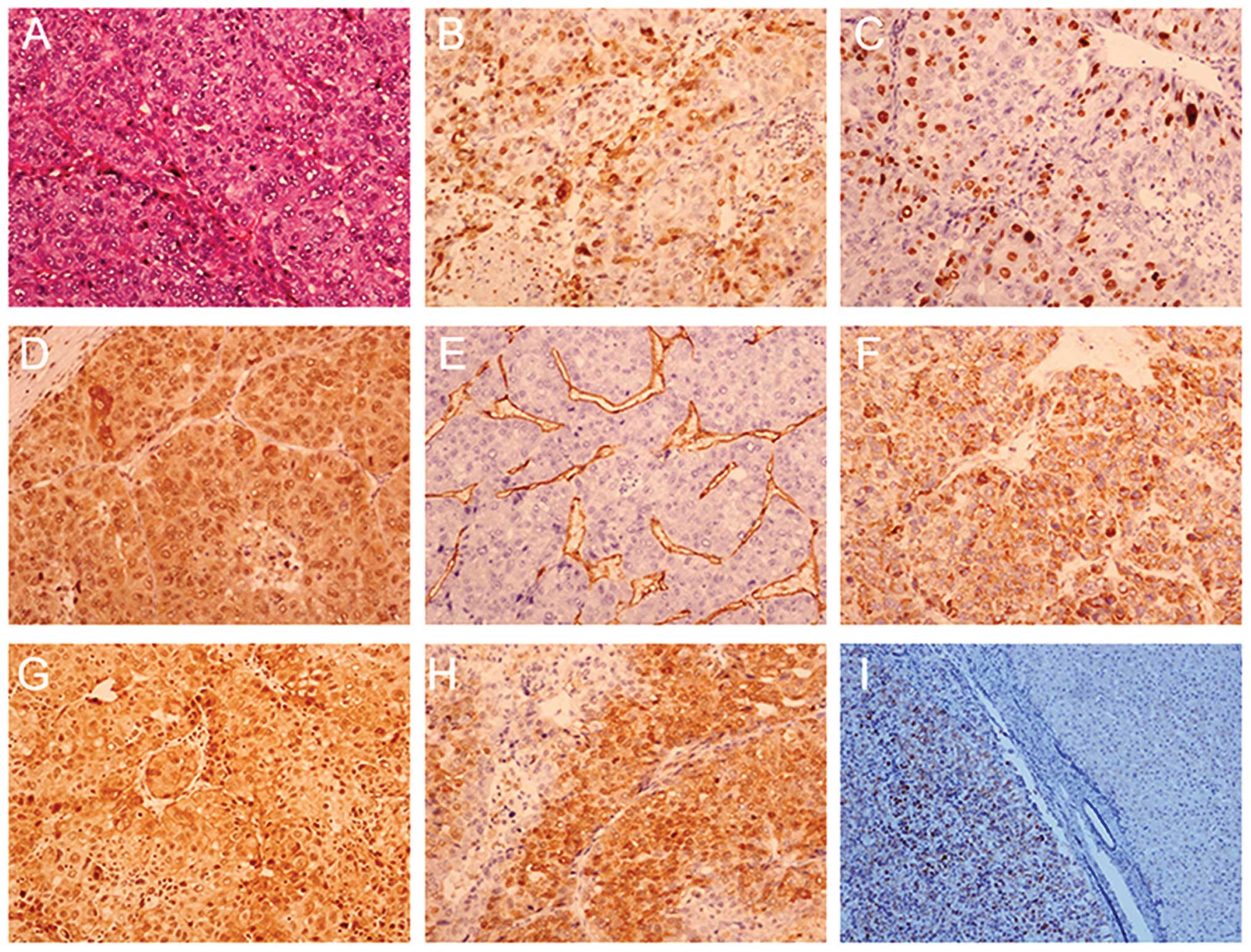
specimens
The expression of histological biomarkers was
examined in the HCC specimens (Fig.
1A–H). Immunohistochemical staining for the CHCHD2 biomarker
was markedly positive and diffuse throughout the tumor tissue
(Fig. 1I, right) and absent from
the adjacent normal liver tissue (Fig.
1I, left).
Identification of the CHCHD2 gene
To elucidate the biological role of HVC NS2, the
present study aimed to identify potential proteins which interact
with NS2. SSH was introduced to establish a subtractive cDNA
library of the HepG2 cells transfected with a pcDNA3.1 (−)−NS2
expression plasmid. To evaluate the efficiency of cDNA subtraction,
the transcriptional expression levels of the housekeeping gene
GAPDH was evaluated by RT-qPCR using the Power SYBR Green PCR Mix
(Applied Biosystems Life Technologies) in the subtracted and
unsubtracted cDNA libraries. The detection of GAPDH sequences for
the two subtractions required 28 PCR cycles with the subtracted
cDNA as the template, whereas only 18 cycles were required to
amplify the GAPDH from the unsubtracted cDNA. Thus, the commonly
expressed gene GAPDH was significantly depleted from the subtracted
cDNA libraries. In total, 30 subtractive cDNA clones were obtained
by performing a BLAST search for comparison with NCBI RefSeq,
GenBank and dbEST. The CHCHD2 gene was successfully cloned from the
HepG2 cell cDNA and was confirmed by sequencing. According to the
NCBI database (http://www.ncbi.nlm.nih.gov/), CHCHD2 (GenBank
accession no. AY605046.1) was named alternatively as coiled-c
oil-helix-coiled-coil-helix domain containing 2. CHCHD2 was located
at 7p11.2, containing 456 nucleotide bases and encoding 151
aas.
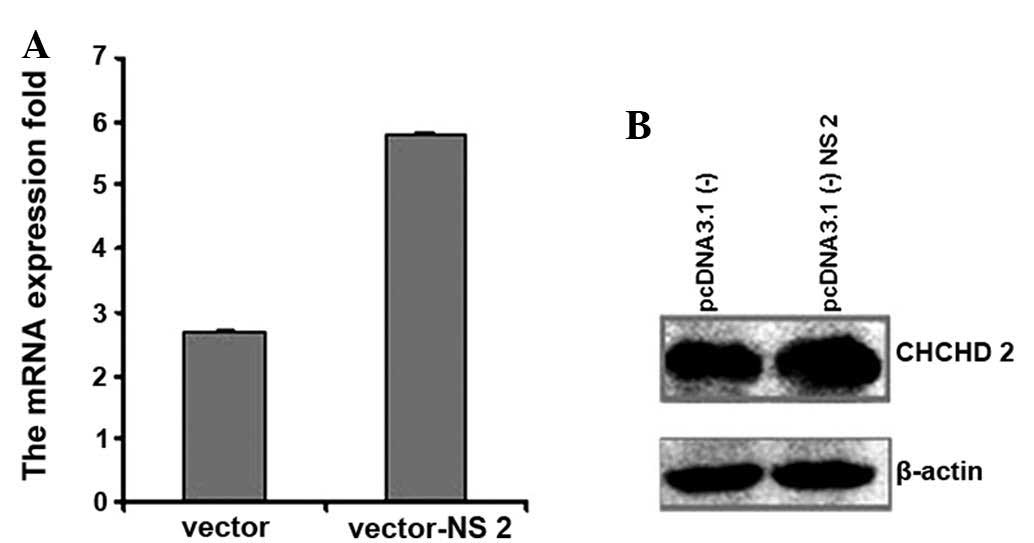
Upregulation of CHCHD2 by HCV NS2
The present study demonstrated that CHCHD2 was
upregulated by HCV NS2 in the SSH. In order to demonstrate whether
HCV NS2 induced the expression of CHCHD2, CHCHD2 mRNA and protein
were analyzed. To determine this, 4 μg pcDNA3.1 (−)−NS2 was
transfected into the HepG2 cells and a pcDNA3.1 (−) vector was used
as a control. An increase in mRNA expression of CHCHD2 was detected
(~2-fold) using RT-qPCR 24 h after the initial transfection
(Fig. 2A). The whole-cell lysates
were then analyzed by western blot analysis with a mouse monoclonal
antibody to detect the protein levels of CHCHD2. Transfection of
the cells with pcDNA3.1 (−)−NS2 caused an increase in the protein
expression of CHCHD2 compared with transfection with the pcDNA3.1
(−) vector (Fig. 2B). These
results suggested that NS2 upregulated the expression of
CHCHD2.
Cloning and analysis of the promoter of
CHCHD2
Using the NCBI database, the present study
characterized the 5′-flanking region upstream of the CHCHD2 gene.
Typical TATA boxes and a high GC content was present, suggesting
the possibility of a number of transcription factor binding sites
within this region. To identify and analyze the promoter of CHCHD2,
a 1,964 bp genomic DNA fragment was amplified from the genomic DNA,
which was previously isolated from the HepG2 cells. A luciferase
assay was then performed by cloning the putative promoter region of
CHCHD2 into the pGL4.10 basic reporter plasmid to produce a
construct termed N1, which was used as a template for further
shortened CHCHD2 promoter constructs. The luciferase assays were
performed 24 h after transfection of the HepG2 cells. Compared with
the pGL4.10 basic plasmid, the luciferase activity of N1 increased
(~27 fold), suggesting that the CHCHD2 promoter was active in the
HepG2 cells. Subsequently, the transcriptional regulatory activity
was determined in the promoter region. An additional three
constructs containing sequentially truncated promoter fragments
were generated (Fig. 3A). As shown
in Fig. 3B, there was a decrease
in activity when the 5′-end was shortened from N1 to N2, suggesting
that the regulatory elements located between −1871 and −1691 may
have acted as potential enhancers. However, there was a loss of
promoter activity with N4. The serial deletion analysis therefore
suggested that N3, including a 350-bp fragment of the promoter
(between −257 and +93), contained the requisite sequences important
for transcriptional activity.
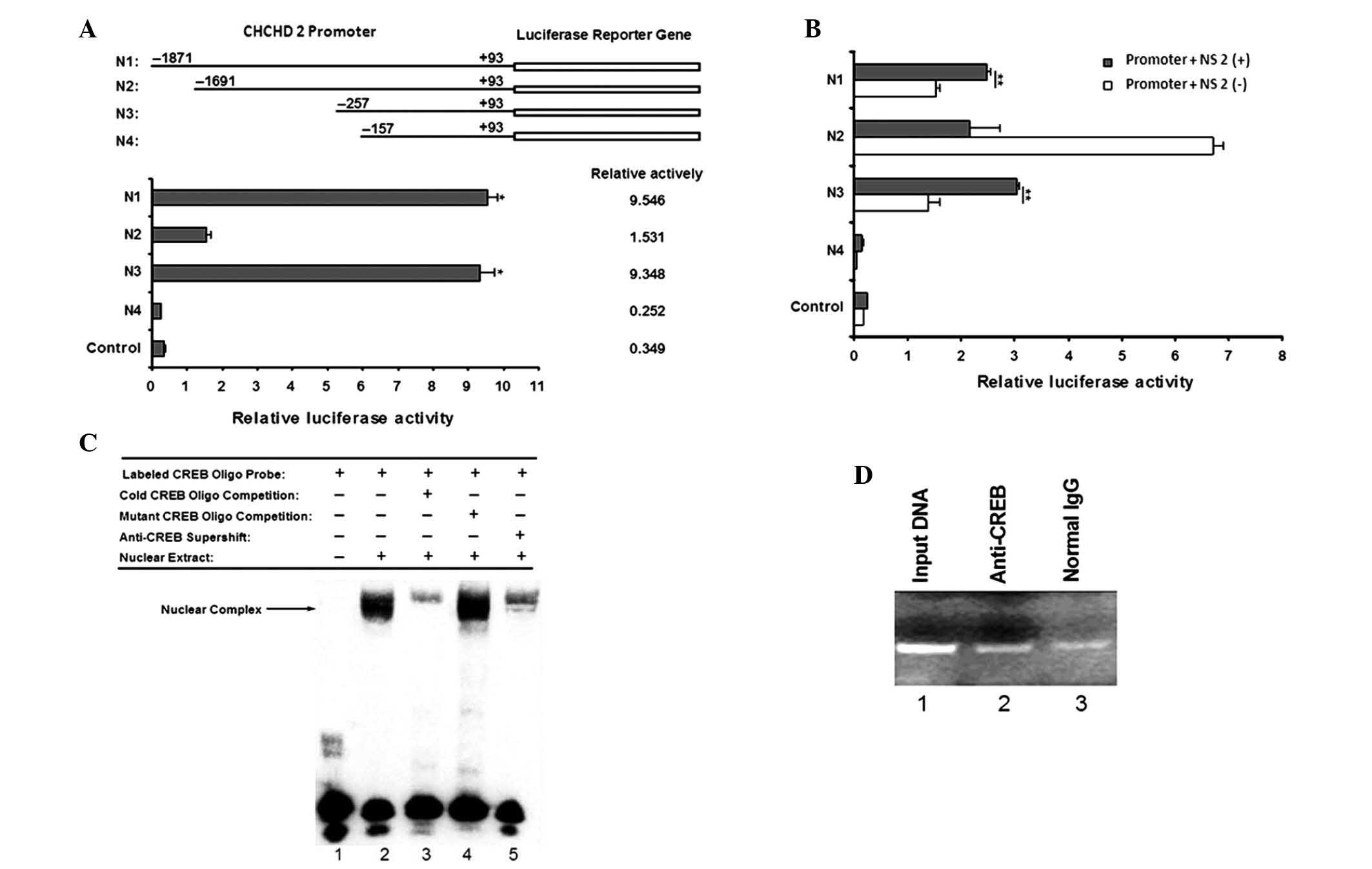 | Figure 3Assessment of the CHCHD2 promoter.
(A) Identification of the proximal CHCHD2 gene promoter. The CHCHD2
gene promoter was constructed and pGL4.10 Basic was used as
control. The ratio of luciferase activity to that of the Renilla
control is shown in the graph. Values are presented as the mean ±
SD of six wells; *P<0.05. (B) Role of the HCV NS2
protein on the CHCHD2 gene promoter. HepG2 cells were transiently
cotransfected with the reconstructed CHCHD2 promoter DNA combined
with pcDNA3.1 (−)−NS2. The pGL4.10 Basic + pcDNA3.1 (−) vector was
used as a control. The data represent the mean ± SD of six wells.
Statistical significance was determined by an unpaired two-tailed
Student's t-test; **P<0.01. (C) CREB bound to the
putative CREB binding site in the CHCHD2 gene promoter. With the
exception of lane 1, the nuclear extracts prepared from HepG2 cells
were incubated with labeled oligo-nucleotide containing the
wild-type CREB binding site under various conditions. The arrow
indicates the DNA/protein complex of the CREB. (D) Binding of CREB
to the minimal CHCHD2 gene promoter was analyzed using a ChIP
assay. Lane 1, PCR product derived from 1% of the
unimmunoprecipitated genomic DNA; lane 2, PCR product derived from
the DNA template immunoprecipitated by the anti-CREB antibody; lane
3, PCR product derived from the DNA template immunoprecipitated by
normal IgG. A band of 159 bp containing the CREB binding site in
the CHCHD2 promoter gene was amplified. The ChIP assay confirmed
the binding of CREB to the CHCHD2 gene promoter DNA. CHCHD2,
coiled-coil-helix-coiled-coil-helix domain containing 2; SD,
standard deviation; HCV, hepatitis C virus; CREB, cyclic adenosine
monophosphate response element-binding protein; ChIP, chromatin
immunoprecipitation; IgG, immunoglobulin G; NS2, nonstructural
protein 2. |
To examine the effect of the HCV NS2 protein on the
CHCHD2 promoter, transient cotransfection was performed using the
pcDNA3.1 (−)−NS2 and CHCHD2 promoters in the HepG2 cells. The
luciferase reporter assay results revealed that the expression of
NS2 promoted the production of luciferase in N1 and N3 compared
with the controls (Fig. 3B),
suggesting that NS2 was responsible for the upregulated gene
expression of CHCHD2 by increasing the transcriptional activity of
its promoter regions between nucleotides −257 and +93.
Specific binding of the CREB
transcription factor to the CHCHD2 proximal promoter
The present study used the Promoter Scan (http://www-bimas.cit.nih.gov/molbio/proscan/) and
TFSEARCH (http://www.cbrc.jp/research/db/TFSEARCH.html)
databases to identify the putative transcription factor binding
sites. Based on the scans for the consensus transcription factor
binding motifs, one major cis-acting element, with the highest
score for CREB, was present within the minimal promoter region
(between −257 and +93). The CREB binding site was identified
between −223 and −216. This suggested that activation of the CHCHD2
promoter by NS2 was regulated by the binding of CREB to the
putative binding site.
To examine whether CREB was able to bind to the
CHCHD2 promoter, EMSAs were performed. The first oligonucleotide
selected for EMSA contained sequences matching a consensus CREB
binding site. The nuclear extracts from the HepG2 cells exhibited
marked binding to a wild-type probe containing a CREB binding site
(Fig. 3C; lane 2). Competition
experiments were then performed using unlabeled probes and mutant
probes (Fig. 3C; lanes 3 and 4).
The DNA-protein complex was specific, as it was markedly
out-competed by a 100-fold excess of the unlabeled probe, whereas
the mutant probe, containing a 4-bp deletion, failed to compete.
However, the nuclear complex likely contained CREB as part of a
larger protein complex, since the complex in lanes 3 and 5
(Fig. 3C) was reduced by the
unlabeled probe and antibody, respectively. These results suggested
that CREB was able to bind to the CHCHD2 minimal promoter.
To assess whether CREB was able to bind to the
CHCHD2 proximal promoter in vitro, ChIP assays followed by
qPCR were performed. The results demonstrated an increase of CREB
on N3 compared with those in the controls, which were precipitated
with IgG (Fig. 3D). A 159-bp band
containing the CREB binding site in the CHCHD2 promoter gene was
amplified following qPCR amplification. By contrast, the IgG
immunoprecipitated products did not contain the CHCHD2 promoter DNA
sequences (Fig. 3D; lane 3). As a
positive control, the isolated genomic DNA input was directly used
for qPCR analysis and the corresponding 159-bp fragment was also
identified (Fig. 3D; lane 1).
These data revealed that CREB can specifically bind to the CHCHD2
promoter.
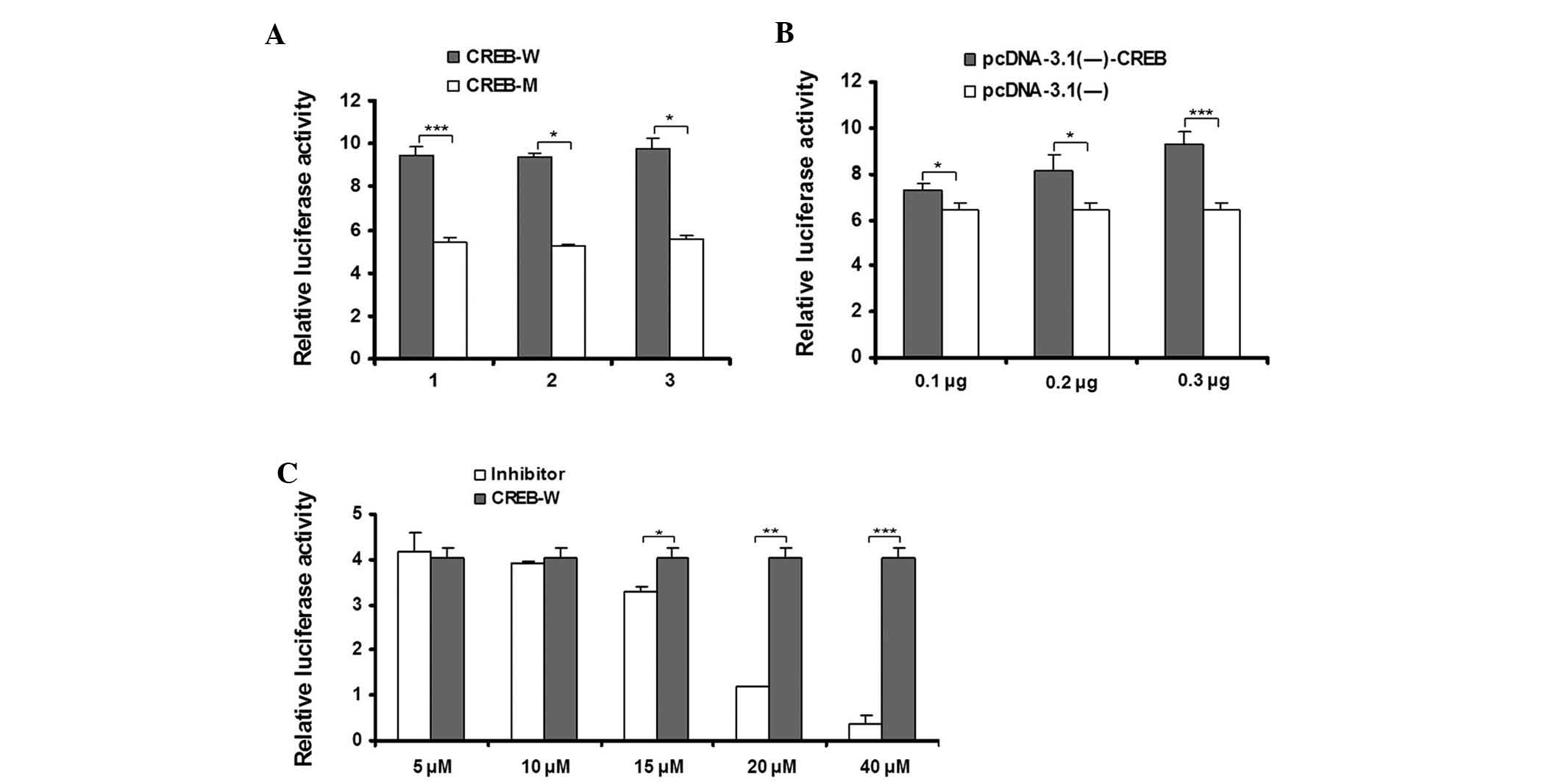
Functional analysis of the effect of CREB
on CHCHD2 promoter activity
To confirm whether the transcription factor CREB has
a functional role in the activation of the CHCHD2 promoter,
site-directed mutagenesis of the CREB binding site within N3 was
performed and the luciferase activity between the wild-type and
CREB mutant binding site were compared using transient transfection
experiments. As shown in Fig. 4A,
The mutations of the CREB site had a marked effect on the promoter
activity, as demonstrated by a 40%-decrease in luciferase activity.
The mutational analyses indicated that abrogation of the CREB
binding site was sufficient to disrupt the activity of the CHCHD2
promoter.
To assess the effect of CREB on the expression of
the CHCHD2 promoter, co-transfection experiments of N3 with an
expression plasmid for CREB were performed. As shown in Fig. 4B, the transcriptional activity of
the CHCHD2 promoter was not altered in the control group. By
contrast, the transcriptional activity was markedly induced by
pcDNA3.1 (−)−CREB. The presence of CREB (between 0.1 and 0.3
μg) induced a dose-dependent increase in luciferase
activity, suggesting the importance of CREB in regulating the gene
expression of CHCHD2.
With respect to PKA, the phosphorylation of CREB was
shown to increase the transcription of cAMP-responsive genes and
the PKA inhibitor H89 has been used to inhibit CREB phosphorylation
(21). Therefore, H89 was used to
evaluate the effect of CREB on the transcriptional activity of
CHCHD2. The inhibitor concentrations were based on previous
studies, which established effective levels, all involving HepG2
cells (22–24). As shown in Fig. 4C, the N3 luciferase activity was
disrupted in the HepG2 cells in the presence of H89. These results
suggested that CREB is important for maintaining the
transcriptional activity of CHCHD2.
Discussion
HCV NS2 is a 217 aa-long cysteine protease, which
cleaves the NS2–3 junction in cooperation with the N-terminal 180
aas of NS3, forming serine-type protease, likely in a rapid
intramolecular reaction (5,25).
NS2 potentially modulates the host cell environment during HCV
infection through interference with gene expression and through the
induction of hepatocellular apoptosis, particularly as CHCHD2
localizes to the mitochondria (26). A potential role for NS2 in the
modulation of the host cell environment has potentially important
implications for the establishment of persistent infection and the
pathogenesis of chronic HCV infection (8,27).
Several HCV proteins have been implicated in the modulation of cell
signaling and apoptosis, including core, E2, NS5A and NS2. The
expression of these proteins in the liver prevents the release of
cytochrome c from the mitochondria and suppresses the activity of
caspase 9 and caspase 3/7 without affecting caspase 8 (28). Therefore, the HCV proteins may be
involved in the mitochondrial intrinsic apoptotic pathway, which
involves increased mitochondrial membrane permeabilization and the
release of pro-apoptotic factors, resulting in cell death (29). Numerous interactions between NS2
and viral or cellular proteins have been reported. To further
examine the function of NS2, the present study used SSH to identify
CHCHD2. At present, no correlation has been observed between CHCHD2
and NS2. The inhibition of NS2-induced apoptosis may, in part, be
due to its ability to induce the expression of CHCHD2, although, to
the best of our knowledge, no previous study has focused on this
possible pathogenic mechanism.
CHCHD2 is connected to 83 genes in the co-expression
network, which are involved in glycolysis and translation (30). Together, the findings of the
present study suggested that CHC H D2 is important in translation
in human cells. Previously, the gene expression of human CHCHD2 has
been determined by transcriptome analysis to be increased in
certain types of cancer tissue compared with that in normal
tissues, suggesting that CHCHD2 may be associated with the
progression of cancer (31,32).
On the basis of a previous study, CHCHD2 is important in enhancing
cell migration-promoting activity (13), instead of promoting cell
proliferation. Mitochondrial oxidative phosphorylation (OxPhos) is
central to energy homeostasis and human health by serving as the
primary generator of ATP in the cell (20,33–36)
and knockdown of CHCHD2 resulted in OxPhos deficits.
In the present study, to investigate the function of
CHCHD2, the mechanism of CHCHD2 transcriptional regulation was
examined. The NS2 transregulation gene was identified using SSH
and, according to RT-qPCR and western blot analyses, NS2 was able
to upregulate the expression of CHCHD2. In the present study, a
region of the CHCHD2 promoter, which was 1,964 bp upstream of the
transcription start site, was cloned, the promoters were deleted,
the plasmid which was reconstructed, was constructed in a stepwise
fashion and analyzed for luciferase activity in the HepG2 cells. A
minimal promoter sequence, spanning between nucleotides −257 and
+93, was sufficient to drive the expression of CHCHD2.
The present study identified a novel gene coding for
the NS2 transregulated protein CHCHD2. Previous studies have
demonstrated that CHCHD2 is a member of a protein family that
contains the (coiled coil 1)-(helix 1)-(coiled-coil 2)-(helix 2)
(CHCH) domain (23); however, no
previous studies have reported an interaction between CHCHD2 and
HCC.
To date, immunohistochemical analysis of the
expression of CHCHD2 has not been performed in large scale studies
of human tumors. The present study identified the overexpression of
CHCHD2 in liver tissues, which was particularly evident in early
and well-differentiated HCC compared with the surrounding
non-cancerous liver tissue. Therefore, these results suggested that
CHCHD2 may be a novel and useful diagnostic biomarker for early
HCC.
Acknowledgments
This study was supported by grants from the Healthy
Talent Leadership Programs of Beijing (no. 2009-1-09), the National
Importance Foundation of Infectious Diseases during the Five-Year
Plan Period of China (nos. 2012ZX10002003, 2013ZX10002005 and
2012ZX10004904), the Science and Technology Planning Project of
Beijing (no. 11320016) and the Specialized Research Fund for the
Doctoral Program of Higher Education (no. 20121107110012).
References
|
1
|
Lohmann V, Koch JO and Bartenschlager R:
Processing pathways of the hepatitis C virus proteins. J Hepatol.
2:11–19. 1996.
|
|
2
|
Grakoui A, Wychowski C, Lin C, Feinstone
SM and Rice CM: Expression and identification of hepatitis C virus
polyprotein cleavage products. J Virol. 67:1385–1395.
1993.PubMed/NCBI
|
|
3
|
Flint M, Thomas JM, Maidens CM, et al:
Functional analysis of cell surface-expressed hepatitis C virus E2
glycoprotein. J Virol. 73:6782–6790. 1999.PubMed/NCBI
|
|
4
|
Welbourn S and Pause A: The hepatitis C
virus NS2/3 protease. Curr Issues Mol Biol. 9:63–69.
2007.PubMed/NCBI
|
|
5
|
Dentzer TG, Lorenz IC, Evans MJ and Rice
CM: Determinants of the hepatitis C virus nonstructural protein 2
protease domain required for production of infectious virus. J
Virol. 83:12702–12713. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oem JK, Jackel-Cram C, Li YP, et al:
Activation of sterol regulatory element-binding protein 1c and
fatty acid synthase transcription by hepatitis C virus
non-structural protein 2. J Gen Virol. 89:1225–1230. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu M, Liu Y, Cheng J, et al:
Transactivating effect of hepatitis C virus core protein: a
suppression subtractive hybridization study. World J Gastroenterol.
10:1746–1749. 2004.PubMed/NCBI
|
|
8
|
Zhang LY, Cheng J, Deng H, Liu Y and Wang
Lin: Cloning and identification of gene NS2TP transregulated by
non-structural protein 2 of hepatitis C virus. World Chin J
Digestol. 13:1700–1704. 2005.
|
|
9
|
Seo M, Lee WH and Suk K: Identification of
novel cell migration-promoting genes by a functional genetic
screen. FASEB J. 24:464–478. 2010. View Article : Google Scholar
|
|
10
|
Hong Y, Lan MD, Wang Q, et al: Prokaryotic
expression and polyclonal antibody preparation of nonstructural
protein 2 transactivated protein of hepatitis C virus. Chin J
Infect Dis. 27:217–220. 2009.
|
|
11
|
Cavallaro G: Genome-wide analysis of
eukaryotic twin CX9C proteins. Mol Biosyst. 6:2459–2470. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Montminy M, Brindle P, Arias J, Ferreri K
and Armstrong R: Regulation of somatostatin gene transcription by
cyclic adenosine monophosphate. Metabolism. 45(8 Suppl 1): 4–7.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shaywitz AJ and Greenberg ME: CREB: a
stimulus-induced transcription factor activated by a diverse array
of extracellular signals. Annu Rev Biochem. 68:821–861. 1999.
View Article : Google Scholar
|
|
14
|
Chrivia JC, Kwok RP, Lamb N, Hagiwara M,
Montminy MR and Goodman RH: Phosphorylated CREB binds specifically
to the nuclear protein CBP. Nature. 365:855–859. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mayr B and Montminy M: Transcriptional
regulation by the phosphorylation-dependent factor CREB. Nat Rev
Mol Cell Biol. 2:599–609. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ravnskjaer K, Kester H, Liu Y, et al:
Cooperative interactions between CBP and TORC2 confer selectivity
to CREB target gene expression. EMBO J. 26:2880–2889. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Johannessen M, Delghandi MP and Moens U:
What turns CREB on? Cell Signal. 16:1211–1227. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
International Working Party: Terminology
of nodular hepatocellular lesions. Hepatology. 22:983–993. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lin CT, Lin CR, Tan GK, Chen W, Dee AN and
Chan WY: The mechanism of Epstein-Barr virus infection in
nasopharyngeal carcinoma cells. Am J Pathol. 150:1745–1756.
1997.PubMed/NCBI
|
|
20
|
Nayak RR, Kearns M, Spielman RS and Cheung
VG: Coexpression network based on natural variation in human gene
expression reveals gene interactions and functions. Genome Res.
19:1953–1962. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Davies SP, Reddy H, Caivano M and Cohen P:
Specificity and mechanism of action of some commonly used protein
kinase inhibitors. Biochem J. 351:95–105. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chang SH, Garcia J, Melendez JA, Kilberg
MS and Agarwal A: Haem oxygenase 1 gene induction by glucose
deprivation is mediated by reactive oxygen species via the
mitochondrial electron-transport chain. Biochem J. 371:877–885.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schaak S, Cayla C, Lymperopoulos A, et al:
Transcriptional down-regulation of the human alpha2C-adrenergic
receptor by cAMP. Mol Pharmacol. 58:821–827. 2000.PubMed/NCBI
|
|
24
|
Citterio C, Jones HD, Pacheco-Rodriguez G,
Islam A, Moss J and Vaughan M: Effect of protein kinase A on
accumulation of brefeldin A-inhibited guanine nucleotide-exchange
protein 1 (BIG1) in HepG2 cell nuclei. Proc Natl Acad Sci USA.
103:2683–2688. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Santolini E, Pacini L, Fipaldini C,
Migliaccio G and Monica N: The NS2 protein of hepatitis C virus is
a transmembrane polypeptide. J Virol. 69:7461–7471. 1995.PubMed/NCBI
|
|
26
|
Hijikata M, Mizushima H, Akagi T, et al:
Two distinct proteinase activities required for the processing of a
putative nonstructural precursor protein of hepatitis C virus. J
Virol. 67:4665–4675. 1993.PubMed/NCBI
|
|
27
|
Dentzer TG, Lorenz IC, Evans MJ and Rice
CM: Determinants of the hepatitis C virus nonstructural protein 2
protease domain required for production of infectious virus. J
Virol. 83:12702–12713. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Papatheodoridis GV, Hadziyannis E,
Tsochatzis E, et al: Serum apoptotic caspase activity in chronic
hepatitis C and nonalcoholic Fatty liver disease. J Clin
Gastroenterol. 44:87–95. 2010. View Article : Google Scholar
|
|
29
|
Kannan RP, Hensley LL, Evers LE, Lemon SM
and McGivern DR: Hepatitis C virus infection causes cell cycle
arrest at the level of initiation of mitosis. J Virol.
85:7989–8001. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jin C, Myers AM and Tzagoloff A: Cloning
and characterization of MRP10, a yeast gene coding for a
mitochondrial ribosomal protein. Curr Genet. 31:228–234. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yanai I, Benjamin H, Shmoish M, et al:
Genome-wide midrange transcription profiles reveal expression level
relationships in human tissue specification. Bioinformatics.
21:650–659. 2005. View Article : Google Scholar
|
|
32
|
Shmueli O, Horn-Saban S, Chalifa-Caspi V,
et al: GeneNote: whole genome expression profiles in normal human
tissues. C R Biol. 326:1067–1072. 2003. View Article : Google Scholar
|
|
33
|
Mootha VK, Bunkenborg J, Olsen JV, et al:
Integrated analysis of protein composition, tissue diversity, and
gene regulation in mouse mitochondria. Cell. 115:629–640. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mayr B and Montminy M: Transcriptional
regulation by the phosphorylation-dependent factor CREB. Nat Rev
Mol Cell Biol. 2:599–609. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Servillo G, Della Fazia MA and
Sassone-Corsi P: Coupling cAMP signaling to transcription in the
liver: pivotal role of CREB and CREM. Exp Cell Res. 275:143–154.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Abramovitch R, Tavor E, Jacob-Hirsch J, et
al: A pivotal role of cyclic AMP-responsive element binding protein
in tumor progression. Cancer Res. 64:1338–1346. 2004. View Article : Google Scholar : PubMed/NCBI
|