Introduction
The inflammatory response is involved in the
pathogenesis of the most common forms of heart disease, including
myocarditis, cardiomyopathy, myocardial infarction and heart
failure (1). Although the initial
inflammatory reaction is a protective response to stressors,
including infection or tissue injury, prolonged inflammation leads
to additional cardiac injury and cardiomyocyte loss (2). Of note, cardiomyocytes themselves are
a significant source of pro-inflammatory cytokines, including tumor
necrosis factor α (TNF-α), interleukin (IL)-1β and IL-6 (3). Toll-like receptor 4 (TLR4), an innate
immune receptor expressed on immune cells and cardiomyocytes, is
critical in the activation of the nuclear factor-κB (NF-κB) pathway
and the induction of inflammatory cytokines within cardiomyocytes
(4). TLR4 signaling is involved in
numerous cardiovascular diseases, including myocardial ischemia,
ischemia/reperfusion, cardiac hypertrophy, cardiomyopathy and heart
failure (5–9). Therefore, pharmacological
interventions which disrupt the TLR4-induced inflammatory response
in cardiomyocytes may be a promising approach for the treatment of
certain cardiovascular diseases.
Icariin (ICA;
C33H40O15; MW, 676.66), a
prenylated flavonol glycoside, is the major active component
isolated from plants of the Epimedium family, which are used
in Traditional Chinese Medicine for the treatment of rheumatism,
osteoporosis and hypogonadism (10,11).
Icariin has an anti-inflammatory effect in a number of tissues and
cells, and a range of pharmacological properties of icariin have
been revealed, including immunoregulation, regulation of oxidative
stress, anti-apoptotic effects and stimulation of angiogenesis
(10,12–14).
Recently, Song et al (13)
showed that icariin attenuated cardiac remodeling in rats with
congestive heart failure through inhibition of matrix
metalloproteinase (MMP) activity and protection of cardiomyocytes
from apoptosis. This suggests that icariin has a cardioprotective
role. However, it has yet to be elucidated whether icariin inhibits
the inflammatory response and subsequent injury in stimulated
cardiomyocytes.
Lipopolysaccharide (LPS), a component of the outer
layer of the gram-negative bacterial cell wall, is an agonist of
TLR4 and has been shown to induce cardiomyocyte dysfunction by
activating TLR4 and promoting the production of inflammatory
mediators (4). In the present
study, LPS was used to induce inflammatory injury in H9c2 rat
cardiomyocytes treated with icariin, and the effects on cell
viability, apoptosis and the production of inflammatory mediators
were investigated. In addition, the possible mechanisms underlying
the effects of icariin on these processes were examined.
Materials and methods
Reagents
Icariin (≥94% purity as determined by high
performance liquid chromatography analysis), LPS and
2′,7′-dichlorofluorescin diacetate (DCFH-DA) were obtained from
Sigma-Aldrich (St. Louis, MO, USA). Dulbecco’s modified Eagle’s
medium (DMEM)/F12, fetal bovine serum (FBS), trypsin, penicillin
and streptomycin were obtained from Gibco-BRL (Invitrogen Life
Technologies, Carlsbad, CA, USA). TRIzol® was obtained
from Invitrogen Life Technologies. Transcriptor First Strand cDNA
Synthesis kit and LightCycler® 480 SYBR Green I Master
mix were obtained from Roche Diagnostics (Basel, Switzerland).
ApopTag® Plus Fluorescein In Situ Apoptosis Detection
kit was obtained from EMD Millipore (Billerica, MA, USA). The
following primary rabbit IgG antibodies were obtained from Cell
Signaling Technology, Inc. (Danvers, MA, USA): GAPDH, P-JNK, T-JNK,
P-IκB, T-IκB, P-P65 and T-P65 (1:1000; incubation overnight with
gentle shaking at 4°C). IRDye 800CW conjugated secondary antibodies
were obtained from LI-COR Biosciences (Lincoln, NE, USA).
H9c2 cardiomyocyte culture
The H9c2 embryonic rat heart-derived cell line was
obtained from the Cell Bank of the Chinese Academy of Sciences
(Shanghai, China). Icariin was dissolved in dimethyl sulfoxide
(DMSO; Sigma-Aldrich) at a concentration of 10 mmol/l for storage
at −20°C. Cells were cultured in DMEM/F12 1:1 medium supplemented
with 10% FBS, penicillin (100 U/ml) and streptomycin (100 mg/ml) in
a humidified incubator with an atmosphere of 5% CO2 at
37°C. Cells were seeded at a density of 1×106 per well
onto six-well culture plates for, mRNA extraction; 5×105
per well onto six-well culture plates, for terminal
deoxynucleotidyl transferase-mediated dUTP nick end-labeling
(TUNEL) analysis; 5×103 cells per well in 96-well plates
for reactive oxygen species (ROS) detection; and 1×107
per well onto culture dishes (100 mm), for protein extraction.
Cells were cultured in serum-free medium for 24 h and pre-treated
with icariin for 12 h prior to LPS stimulation.
Cell viability
Cell viability was investigated using a Cell
Counting kit-8 (CCK-8; Sigma-Aldrich) assay. Following icariin
treatment for 12 h, 10 μl CCK-8 solution was added to each
well of the 96-well plate prior to a further 4 h of incubation.
Absorbance was measured at 450 nm using a microplate reader
(Synergy™ HT, BioTek Instruments, Inc., Winooski, VT, USA). The
percentage of cell viability was calculated according to the
formula: Cell viability(%) = optical density (OD) of treatment
group/OD of control group ×100.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RT-qPCR was used to detect the mRNA expression
levels of inflammatory markers, including TNF-α, IL-1β and IL-6, as
described previously (15).
Following 1 h pre-treatment with icariin, H9c2 cells were incubated
with LPS for 12 h prior to the extraction of total RNA using
TRIzol, and their yields and purities were spectrophotometrically
estimated using the A260/A280 and A230/260 ratios via a SmartSpec
Plus Spectrophotometer (Bio-Rad Laboratories, Hercules, CA, USA).
RNA (2 μg from each sample) was reverse-transcribed into
cDNA using oligo (DT) primers (Sangon Biotech Co., Ltd., Shanghai,
China) and the Transcriptor First Strand cDNA Synthesis kit. The
primer sequences used were as follows: GAPDH, F
5′-GACATGCCGCCTGGAGAAAC-3′ and R 5′-AGCCCAGGATGCCCTTTAGT-3′; TNF-α,
F 5′-AGCATGATCCGAGATGTGGAA-3′ and R 5′-TAGACAGAAGAGCGTGGTGGC-3′;
IL-1β, F 5′-GGGATGATGACGACCTGCTAG-3′ and R
5′-ACCACTTGTTGGCTTATGTTCTG-3′; IL-6, F 5′-GTTGCCTTCTTGGGACTGATG-3′
and R 5′-ATACTGGTCTGTTGTGGGTGGT-3′; Bax, F
5′-AAACTGGTGCTCAAGGCCCT-3′ and R 5′-AGCAGCCGCTCACGGAG-3′; Bcl-2, F
5′-CCGGGAGAACAGGGTATGATAA-3′ and R 5′-CCCACTCGTAGCCCCTCTG-3′. PCR
amplifications were quantified using the LightCycler 480 SYBR Green
I Master mix. GAPDH was used as the internal control. The PCR
cycling conditions were as follows: Initial activation at 95°C for
10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 1
min.
TUNEL staining
A TUNEL assay was performed to label apoptotic
nuclei according to the manufacturer’s instructions (ApopTag Plus
Fluorescein In Situ Apoptosis Detection kit) (16). Briefly, following 1 h pre-treatment
with icariin, cells were incubated with LPS for 12 h and
subsequently fixed on coverslips in 1% paraformaldehyde in
phosphate-buffered saline (both from Sinopharm Chemical Reagent
Co., Ltd., Shanghai, China), stained with TUNEL reagents (EMD
Millipore) and DAPI (Invitrogen Life Technologies) and observed
under a microscope (BX51; Olympus Corp., Tokyo, Japan). The index
of cell apoptosis was calculated as the percentage of apoptotic
nuclei/total number of nuclei.
ROS detection
Intracellular ROS generation was determined by
2′,7′-DCFH-DA which is oxidized to fluorescent DCF by ROS.
Following 1 h pre-treatment with icariin, cells were incubated with
LPS for 30, 60 or 120 min. Subsequently, H9c2 cells were washed
twice and incubated with 5 μM DCFH-DA solution in serum-free
medium at 37°C for 30 min in the dark. Data were then collected
using a fluorescence reader (Synergy HT, BioTek Instruments, Inc.)
at an excitation/emission wavelength of 485/530 nm. A fluorescence
microscope (BX51; Olympus Corp.) was also used to evaluate the DCF
fluorescence of cells on coverslips.
Western blot analysis
Western blotting was performed as described
previously (17). Following 1 h
pre-treatment with icariin, cells were incubated with LPS for 2 h.
Cells were subsequently lysed in radioimmunoprecipitation assay
lysis buffer (Guge Biological Technology Co., Wuhan, China), and
the protein concentration was measured using a bicinchoninic acid
protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA) by a
microplate reader (Synergy HT, BioTek Instruments, Inc.). Cell
lysates (50 μg) were electrophoresed by 10% SDS-PAGE,
transferred onto Immobilon-FL transfer membranes (EMD Millipore),
blocked with 5% non-fat milk and incubated with specific primary
antibodies overnight at 4°C prior to incubation with IRDye
800CW-conjugated secondary antibodies. The blots were scanned using
a two-color infrared imaging system (Odyssey, LI-COR
Biosciences).
Statistical analysis
Values are presented as the mean ± standard error of
the mean. Differences among the groups were determined by two-way
analysis of variance followed by a post hoc Tukey test. Comparisons
between two groups were performed using the unpaired Student’s
t-test. P<0.05 was considered to indicate a statistically
significantly difference.
Results
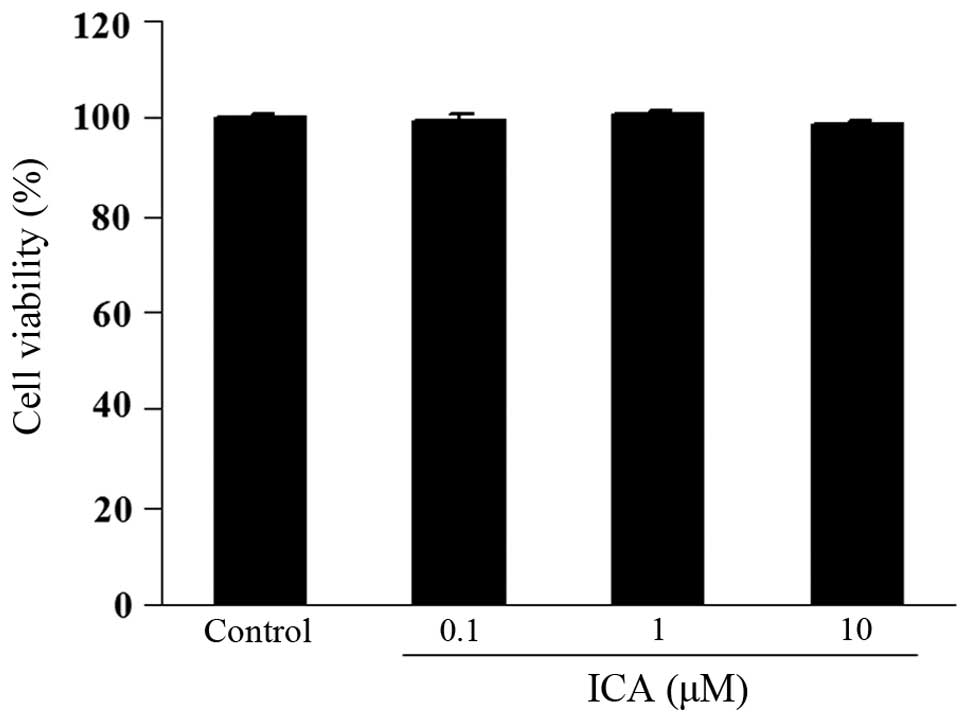
Effect of icariin on cell viability
The potential cytotoxicity of icarrin was examined
using a CCK-8 assay. H9c2 cells were incubated with varying
concentrations of icariin (0.1, 1 and 10 μM) for 12 h. Cell
viability in icariin-treated cells was not significantly different
compared with that of control cells, indicating that icariin (0.1,
1 and 10 μM) did not cause cytotoxicity in H9c2 cells
(Fig. 1).
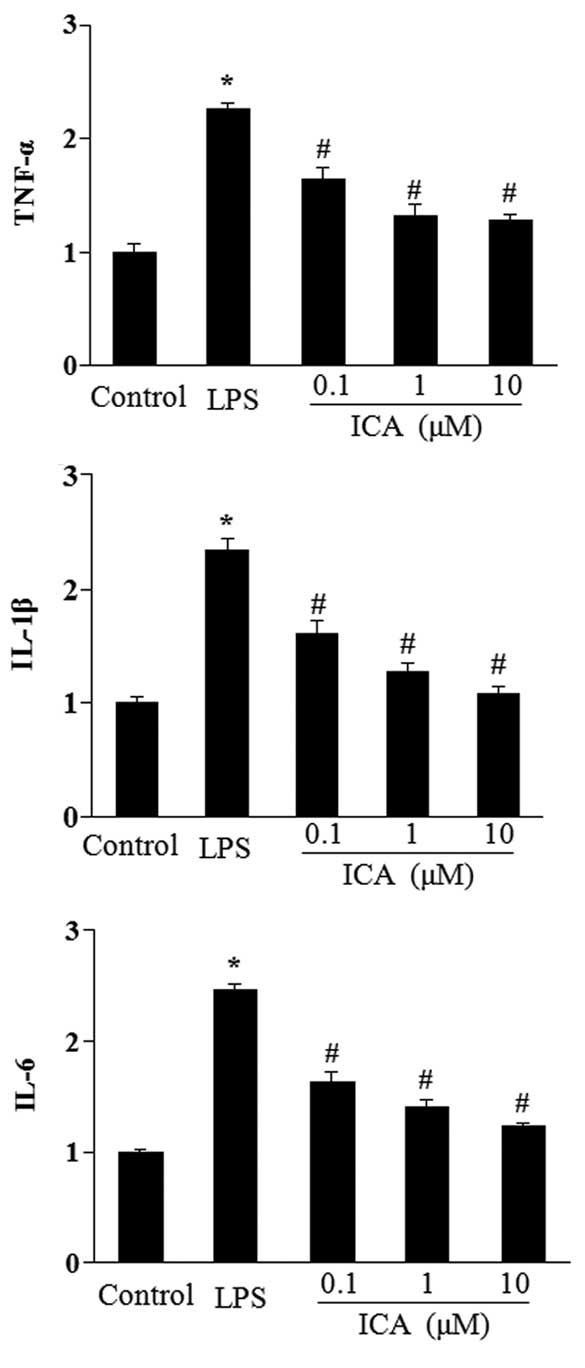
Icariin inhibits the expression of
inflammatory genes induced by LOS in H9c2 cells
The effect of icariin at different concentrations
(0.1–10 μM) on the induction of TNF-α, IL-1β and IL-6 in
response to LPS was measured. Stimulation with LPS for 12 h
significantly increased the mRNA levels of TNF-α, IL-1β and IL-6 in
H9c2 cells. In turn, icariin treatment significantly attenuated
this increase in a concentration-dependent manner (Fig. 2).
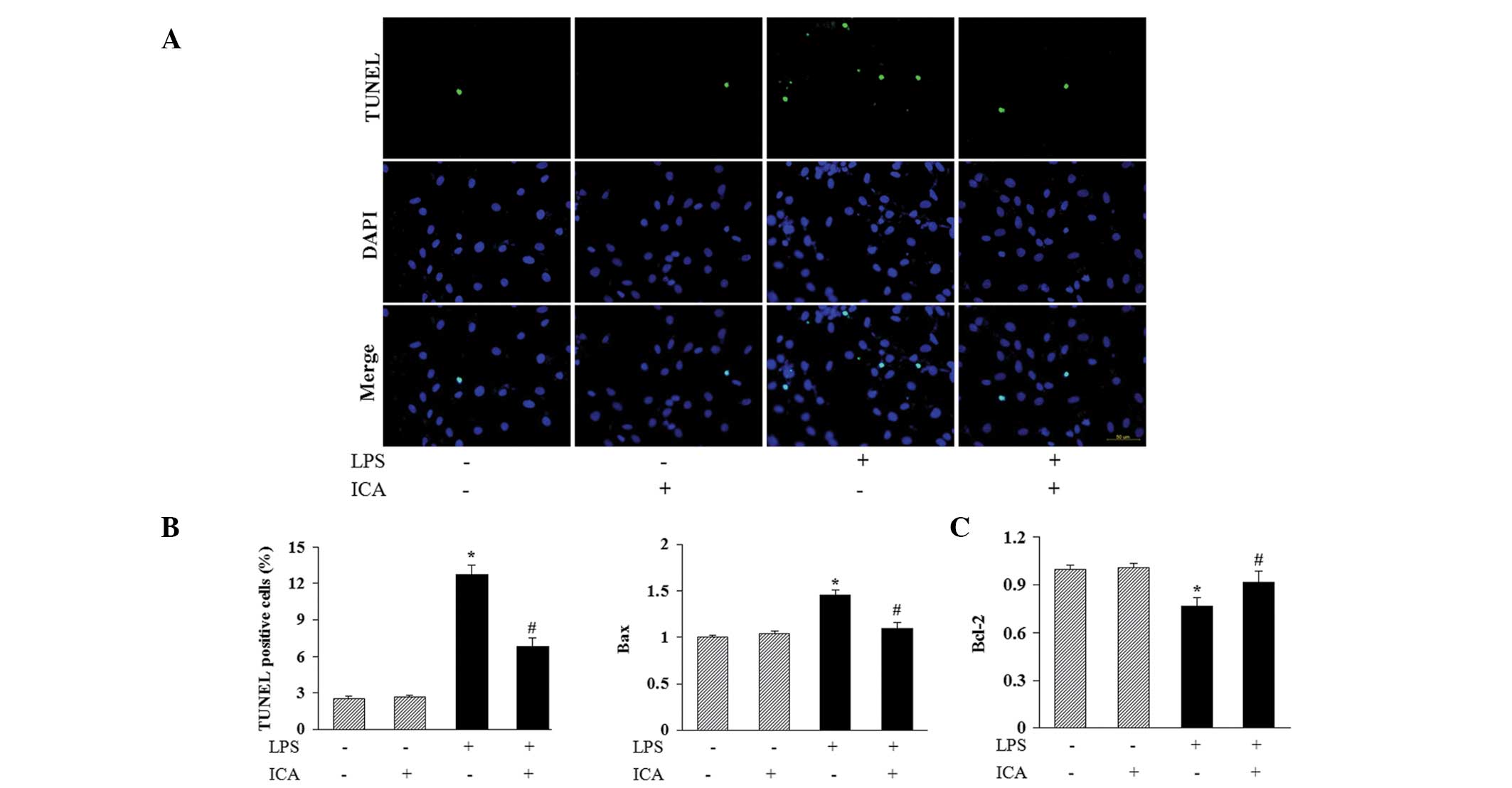
Icariin attenuates LPS-induced apoptosis
in H9c2 cells
To investigate the possible protective role of
icariin in moderating LPS-induced apoptosis of H9c2 cells, TUNEL
staining was used to identify apoptotic nuclei. A significant
increase in the number of TUNEL-positive nuclei was observed in
cells incubated with LPS, and icariin treatment markedly reduced
this LPS-induced cell apoptosis (Fig.
3A and B). In addition, icariin decreased the levels of
expression of B-cell-lymphoma (Bcl-2)-associated X (Bax) mRNA,
while increasing the Bcl-2 mRNA expression levels in H9c2 cells
following LPS stimulation (Fig.
3C). This mechanism may in part mediate the anti-apoptotic
effect of icariin.
Icariin decreases LPS-induced production
of ROS
Cells incubated with DCFH-DA exhibited increased
intensity of fluorescence when treated with LPS, indicating that
LPS induced ROS production in a time-dependent manner (Fig. 4A). Icariin treatment significantly
reduced LPS-induced ROS production at the time-points indicated in
Fig. 4A. In addition, microscopic
examination of DCF-derived fluorescence also suggested that icariin
inhibited the accumulation of intracellular ROS in LPS-treated
cells, which was in accordance with the results obtained with the
fluorescence reader (Fig. 4B).
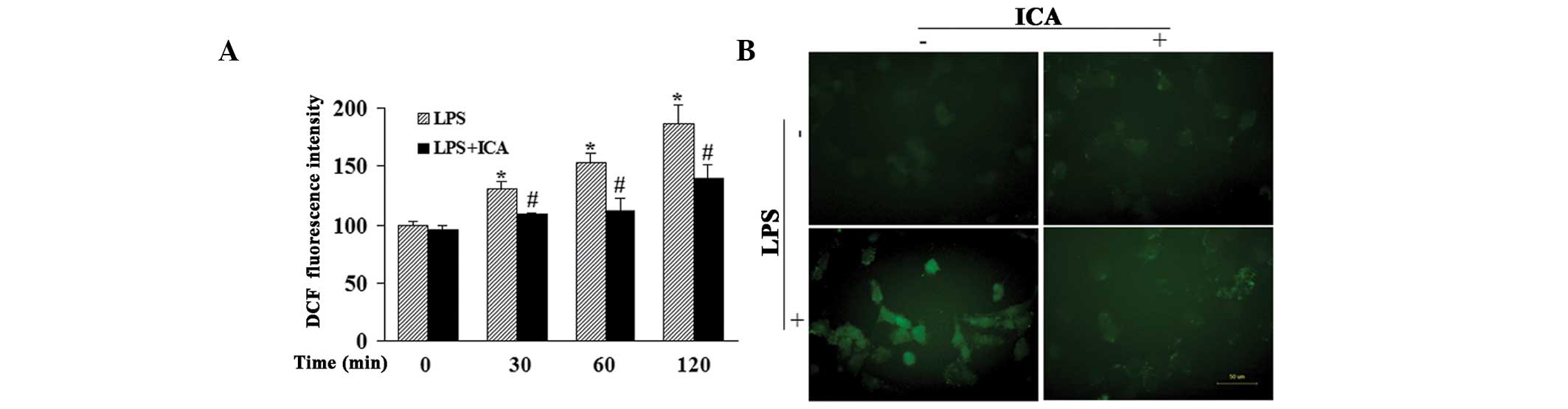 | Figure 4Effect of ICA on ROS production. (A)
Effect of ICA (10 μM) on LPS-induced ROS production at the
indicated time-points, detected by a fluorescence reader. (B)
Effect of ICA (10 μM) on DCF-derived fluorescence in
LPS-treated cells, detected using a fluorescence microscope
(magnification, ×400). *P<0.05, compared with cells
at the 0 h time-point and #P<0.05, compared with
cells treated with LPS at the same time-point. ICA, icariin; ROS,
reactive oxygen species; LPS, lipopolysaccharide; DCF,
2′,7′-dichlorofluorescein. |
Icariin reduces the activation of c-Jun
N-terminal kinase (JNK) and NF-κB in response to LPS
To further investigate the mechanisms underlying the
anti-inflammatory and anti-apoptotic effects of icariin on
LPS-treated H9c2 cells, western blot analysis was used to detect
the activation of JNK and NF-κB, which are key mediators in the
cardiac inflammatory response. The results showed that the
phosphorylated levels of JNK were significantly elevated by LPS,
and that icariin treatment significantly inhibited this LPS-induced
phosphorylation of JNK (Fig. 5A
and B). Furthermore, icariin reduced the phosphorylation and
degradation of IκB in H9c2 cells that occurred in response to LPS
administration, and subsequently decreased the nuclear
translocation and phosphorylated level of NF-κB p65 (Fig. 5C–E).
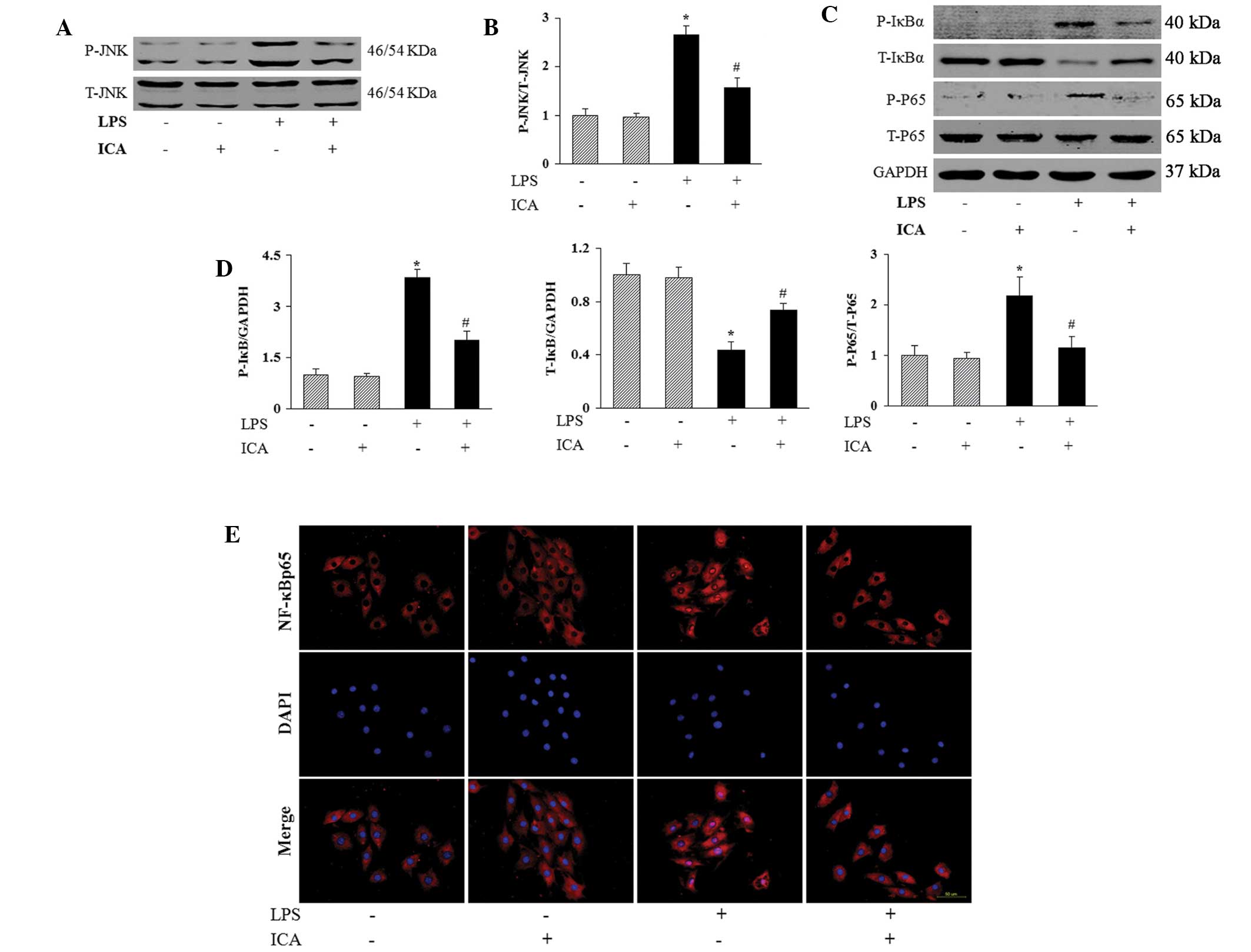 | Figure 5Effect of ICA on the activation of the
JNK and NF-κB pathways. (A) and (B) Effect of ICA on the
phosphorylated levels of JNK in response to LPS (1 μg/ml for
2 h). (C) and (D) Effect of ICA on the phosphorylation and
degradation of IκB in H9c2 cells in response to LPS, and on the
phosphorylated level of NF-κB p65. (A) and (C) Representative
western blots. (B) and (D) Quantitative results. (E) Effect of ICA
on the LPS-induced nuclear translocation of NF-κB p65
(magnification, ×400). *P<0.05, compared with control
cells and #P<0.05, compared with cells treated with
LPS. ICA, icariin; JNK, c-Jun N-terminal kinase; IκB, inhibitor of
kappa B; NF-κB, nuclear factor-κB; LPS, lipopolysaccharide; T,
total; P, phosphorylated. |
Discussion
The present study demonstrated a novel role of
icariin, a prenylated flavonol glycoside isolated from plants of
the Epimedium family, in the protection of cardiomyocytes
from the LPS-induced inflammatory response. Icariin also attenuated
LPS-induced apoptosis in cardiomyocytes in association with the
downregulation of Bax and the upregulation of Bcl-2. The protective
effect of icariin on LPS-stimulated cardiomyocytes may be mediated
by the alleviation of ROS production and inhibition of JNK
activation and the NF-κB signaling cascade.
A previous study showed that the inflammatory
response induced by LPS in cardiomyocytes includes the initial
induction of ROS, which leads to the activation of intracellular
signaling pathways and transcription factors, and subsequently
induces the production of inflammatory mediators, including TNF-α,
IL-1β and IL-6, and the apoptotic response (18,19).
These pro-inflammatory cytokines are involved in the depression of
cardiac function and the progression from cardiac injury to failure
(20). Therefore, blocking
inflammatory signaling may produce beneficial effects in the
dysfunctional heart (21).
Epimedium has been used in Traditional
Chinese Medicine for the treatment of autoimmune disorders, such as
rheumatism, for thousands of years, and its major active component,
icariin, has an anti-inflammatory effect in certain tissues and
cells (10,11). Xu et al (10) showed that pretreatment with icariin
decreased the expression of TNF-α, IL-6, cycloxygenase-2 and
prostaglandin E2 in the lungs of LPS-challenged mice. In addition,
Zeng et al (22)
demonstrated that icariin inhibited the release of TNF-α, IL-1β and
IL-6 in cultured microglia that had been treated with LPS. The
present study showed that icariin downregulated the expression of
TNF-α, IL-1β and IL-6 in LPS-stimulated H9c2 cells, indicating an
anti-inflammatory effect of icariin in cardiomyocytes.
Inflammatory mediators may cause cardiac
cytotoxicity and lead to cardiomyocyte loss through the induction
of apoptotic pathways, thereby promoting cardiac dysfunction
(23). Cardiomyocyte apoptosis is
important in a number of cardiovascular diseases, including cardiac
hypertrophy, heart failure, diabetic cardiomyopathy,
ischemia/reperfusion injury, atherosclerosis and sepsis-associated
cardiac dysfunction (19). The
present study showed that icariin attenuated LPS-induced
cardiomyocyte apoptosis, indicating a potential therapeutic role of
LPS in heart disease. The balance between the regulation of
pro-apoptotic (for example, Bax) and anti-apoptotic (for example,
Bcl-2) proteins determines whether cells undergo apoptosis or
survive. Icariin treatment downregulated the expression of Bax,
while upregulating that of Bcl-2, in LPS-stimulated H9c2 cells.
These changes may contribute to the anti-apoptotic effect of
icariin.
As the results demonstrated that icariin attenuated
the LPS-induced inflammatory response and apoptosis in H9c2 cells,
the mechanisms underlying these beneficial effect were further
investigated. In response to LPS, ROS generation is known to be
markedly elevated, and is involved in the activation of signaling
pathways and the production of inflammatory mediators (18). Previous studies have shown that
icariin protects human umbilical vein endothelial cells from
H2O2-induced apoptosis (24) and inhibits ROS production in
LPS-treated microglia (22). These
finding indicated that icariin has anti-oxidative effects. Studies
have demonstrated that ROS production contributes to the
LPS-induced activation of JNK, which belongs to the
mitogen-activated protein kinase family and is involved in the
induction of the inflammatory response and apoptosis (25,26).
The present study showed that icariin decreased ROS production and
blocked the phosphorylation of JNK in LPS-treated H9c2 cells. The
NF-κB/Rel family of transcription factors are key molecules that
participate in the regulation of the inflammatory response, as well
as certain other cellular processes, including cell growth,
proliferation and apoptosis (27,28).
Inactive NF-κB dimers (classically p65/p50) bind to cytosolic
inhibitory proteins, IκBs. Following stimulation of the NF-κB
pathway, IκBs are phosphorylated, causing their ubiquitination and
degradation, and the subsequent release and activation of NF-κB
(29). It has been reported that
the NF-κB pathway may serve as a target of JNK (25). The present study suggested that
icariin treatment inhibited the phosphorylation and degradation of
IκBs and blocked the subsequent nuclear translocation and
phosphorylation of NF-κB p65 in response to LPS. This indicated
that icariin may inhibit the NF-κB pathway in association with the
downregulation of ROS production and JNK activation.
In conclusion, the present study demonstrated for
the first time, to the best of our knowledge, the alleviating
effect of icariin on the LPS-induced inflammatory response and
apoptosis in cardiomyocytes. This effect may be mediated by
inhibition of the ROS-dependent JNK/NF-κB pathways. These results
provide evidence for the potential application of icariin in the
treatment of inflammatory injury in cardiovascular diseases.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant nos. 81270303 and 81300070) and
the Fundamental Research Funds for the Central Universities of
China (grant no. 2012302020212).
References
|
1
|
Coggins M and Rosenzweig A: The fire
within: cardiac inflammatory signaling in health and disease. Circ
Res. 110:116–125. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hohensinner PJ, Niessner A, Huber K,
Weyand CM and Wojta J: Inflammation and cardiac outcome. Curr Opin
Infect Dis. 24:259–264. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Atefi G, Zetoune FS, Herron TJ, et al:
Complement dependency of cardiomyocyte release of mediators during
sepsis. FASEB J. 25:2500–2508. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Avlas O, Fallach R, Shainberg A, Porat E
and Hochhauser E: Toll-like receptor 4 stimulation initiates an
inflammatory response that decreases cardiomyocyte contractility.
Antioxid Redox Signal. 15:1895–1909. 2011. View Article : Google Scholar
|
|
5
|
Fallach R, Shainberg A, Avlas O, et al:
Cardiomyocyte Toll-like receptor 4 is involved in heart dysfunction
following septic shock or myocardial ischemia. J Mol Cell Cardiol.
48:1236–1244. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hua F, Ha T, Ma J, et al: Protection
against myocardial ischemia/reperfusion injury in TLR4-deficient
mice is mediated through a phosphoinositide 3-kinase-dependent
mechanism. J Immunol. 178:7317–7324. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Satoh M, Nakamura M, Akatsu T, Shimoda Y,
Segawa I and Hiramori K: Toll-like receptor 4 is expressed with
enteroviral replication in myocardium from patients with dilated
cardiomyopathy. Lab Invest. 84:173–181. 2004. View Article : Google Scholar
|
|
8
|
Frantz S, Kobzik L, Kim YD, et al: Toll4
(TLR4) expression in cardiac myocytes in normal and failing
myocardium. J Clin Invest. 104:271–280. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ha T, Li Y, Hua F, et al: Reduced cardiac
hypertrophy in toll-like receptor 4-deficient mice following
pressure overload. Cardiovasc Res. 68:224–234. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu CQ, Liu BJ, Wu JF, et al: Icariin
attenuates LPS-induced acute inflammatory responses: involvement of
PI3K/Akt and NF-kappaB signaling pathway. Eur J Pharmacol.
642:146–153. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu J, Zhou J, Chen X, et al: Attenuation
of LPS-induced inflammation by ICT, a derivate of icariin, via
inhibition of the CD14/TLR4 signaling pathway in human monocytes.
Int Immunopharmacol. 12:74–79. 2012. View Article : Google Scholar
|
|
12
|
Li WW, Gao XM, Wang XM, Guo H and Zhang
BL: Icariin inhibits hydrogen peroxide-induced toxicity through
inhibition of phosphorylation of JNK/p38 MAPK and p53 activity.
Mutat Res. 708:1–10. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Song YH, Cai H, Gu N, Qian CF, Cao SP and
Zhao ZM: Icariin attenuates cardiac remodelling through
down-regulating myocardial apoptosis and matrix metalloproteinase
activity in rats with congestive heart failure. J Pharm Pharmacol.
63:541–549. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chung BH, Kim JD, Kim CK, et al: Icariin
stimulates angiogenesis by activating the MEK/ERK- and
PI3K/Akt/eNOS-dependent signal pathways in human endothelial cells.
Biochem Biophys Res Commun. 376:404–408. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou H, Bian ZY, Zong J, et al: Stem cell
antigen 1 protects against cardiac hypertrophy and fibrosis after
pressure overload. Hypertension. 60:802–809. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou H, Yang HX, Yuan Y, et al:
Paeoniflorin attenuates pressure overload-induced cardiac
remodeling via inhibition of TGFβ/Smads and NF-κB pathways. J Mol
Histol. 44:357–367. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou H, Shen DF, Bian ZY, et al:
Activating transcription factor 3 deficiency promotes cardiac
hypertrophy, dysfunction, and fibrosis induced by pressure
overload. PLoS One. 6:e267442011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pan LL, Liu XH, Gong QH and Zhu YZ:
S-Propargyl-cysteine (SPRC) attenuated lipopolysaccharide-induced
inflammatory response in H9c2 cells involved in a hydrogen
sulfide-dependent mechanism. Amino Acids. 41:205–215. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dong M, Hu N, Hua Y, et al: Chronic Akt
activation attenuated lipopolysaccharide-induced cardiac
dysfunction via Akt/GSK3β-dependent inhibition of apoptosis and ER
stress. Biochim Biophys Acta. 1832:848–863. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
González A, Ravassa S, Beaumont J, López B
and Díez J: New targets to treat the structural remodeling of the
myocardium. J Am Coll Cardiol. 58:1833–1843. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Verma SK, Krishnamurthy P, Barefield D, et
al: Interleukin-10 treatment attenuates pressure overload-induced
hypertrophic remodeling and improves heart function via signal
transducers and activators of transcription 3-dependent inhibition
of nuclear factor-κB. Circulation. 126:418–429. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zeng KW, Fu H, Liu GX and Wang XM: Icariin
attenuates lipopolysaccharide-induced microglial activation and
resultant death of neurons by inhibiting TAK1/IKK/NF-kappaB and
JNK/p38 MAPK pathways. Int Immunopharmacol. 10:668–678. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mann DL: Inflammatory mediators and the
failing heart: past, present, and the foreseeable future. Circ Res.
91:988–998. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang YK and Huang ZQ: Protective effects
of icariin on human umbilical vein endothelial cell injury induced
by H2O2 in vitro. Pharmacol Res. 52:174–182. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tien YC, Lin JY, Lai CH, et al: Carthamus
tinctorius L. prevents LPS-induced TNFalpha signaling activation
and cell apoptosis through JNK1/2-NFkappaB pathway inhibition in
H9c2 cardiomyoblast cells. J Ethnopharmacol. 130:505–513. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ceylan-Isik AF, Zhao P, Zhang B, Xiao X,
Su G and Ren J: Cardiac overexpression of metallothionein rescues
cardiac contractile dysfunction and endoplasmic reticulum stress
but not autophagy in sepsis. J Mol Cell Cardiol. 48:367–378. 2010.
View Article : Google Scholar :
|
|
27
|
Jones WK, Brown M, Ren X, He S and
McGuinness M: NF-kappaB as an integrator of diverse signaling
pathways: the heart of myocardial signaling? Cardiovasc Toxicol.
3:229–254. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu Q, Chen Y, Auger-Messier M and
Molkentin JD: Interaction between NFκB and NFAT coordinates cardiac
hypertrophy and pathological remodeling. Circ Res. 110:1077–1086.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hayden MS and Ghosh S: Shared principles
in NF-kappaB signaling. Cell. 132:344–362. 2008. View Article : Google Scholar : PubMed/NCBI
|