Introduction
Lung cancer is becoming the leading cause of
cancer-associated mortality worldwide, particularly in China
(1,2). For patients with lung cancer,
resistance to therapy is a common phenomenon, which threatens the
success of the treatment currently used against the disease.
Therefore, novel theraperutic strategies are required for the
overcome tumor evasion.
Flavonoids are known for their wide spectrum of
pharmacological properties, including antioxidant, antimicrobial
and anticancer effects (3).
Luteolin (3′,4′,5,7-tetra-hydroxyflavone) is a common dietary
flavanoid, which, similar to several other flavanoids, exists in
several traditional Chinese medicines (4). Luteolin has been demonstrated to
exhibit anticancer properties, including the induction of apoptosis
and cell cycle arrest, and the inhibition of metastasis and
angiogenesis, in several cancer cell lines, including the A549
non-small lung cancer cell (5).
Sirt1 is a well-known NAD+-dependent
class III protein deacetylase, which belongs to the silent
information regulator family (6).
This family has multiple functions and is critically involved in
the stress responses, cellular metabolism and aging, through the
deacetylation of a variety of substrates, including p53,
forkhead-box transcription factors, PGC-1α, NF-κB, Ku70 and
histones (7,8). Sirt1 negatively regulates the tumor
suppressor p53 and other tumor suppressors (9) and inhibits the transcription activity
of AP-1 by targeting c-JUN (10).
However, the possible roles of SIRT1 in the regulation of the
NCI-H460 human lung carcinoma cell apoptosis have not been
reported. The present study investigated the anticancer effect of
luteolin on NCI-H460 by SIRT1 on the regulation of cell apoptosis.
This finding provides novel insight into the mechanisms of
luteolin's anti-lung cancer effects.
Materials and methods
Reagents
Luteolin was obtained from Sigma-Aldrich (St. Louis,
MO, USA) and was dissolved in dimethyl sulfoxide (DMSO;
Sigma-Aldrich) and adjusted to the final concentrations (20, 40, 80
and 160 µM) using complete RPMI-1640 medium. Paclitaxel (Taxol) was
purchased from Haikou Pharmaceutical Factory Co., Ltd., (Haikou,
China). The Taxol was diluted in serum-free culture media and was
administered to cells at a final concentration of 300 nM. Fetal
bovine serum (FBS), RPMI-1640 medium, Dulbecco's modified Eagle's
medium (DMEM) and penicillin-streptomycin were purchased from Gibco
Life Technologies (Grand Island, NY, USA). The
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphnyl-2H-tet-razolium bromide
(MTT) was obtained from Sigma-Aldrich. The primary and secondary
antibodies used in the present study were obtained from Abcam
(Cambridge, UK). All other reagents used were commercially
available and of analytical grade.
Cell lines and cell culture
The NCI-H460 human lung carcinoma cell line and
HEK-293T cell line were obtained from American Type Culture
Collection (Manassas, VA, USA) and cultured in RPMI-1640 and DMEM
culture medium, supplemented with 10% heat-inactivated FBS, 100
U/ml penicillin and 100 µg/ml streptomycin at 37°C in a 5%
CO2 incubator, respectively.
MTT cell viability assay
Cell proliferation was determined using an MTT
assay. Briefly, 1×104 cells were seeded into 96 well
plates and were treated with 0, 20, 40, 80 or 160 µM luteolin for
24 h at 37°C. Following treatment, the medium was replaced with
fresh culture medium, containing 0.5 mg/ml MTT, and incubated for 4
h at 37°C. The culture supernatant was removed and the formazan
crystals were dissolved in 150 µl DMSO for 10 min at room
temperature. The absorbance was measured at 590 nm using an MK3
microplate reader (Thermo Fisher Scientific, Waltham, MA, USA).
Wound healing assay
The NCI-H460 cells were seeded into a 6-well plate
at 2×105 cells/well and cultured until they reached 90%
confluence. A single scratch wound was created on the confluent
monolayers using a micropipette tip (1 mm), which touched the
plate, as previously described (11). The wounded monolayers were washed
with phosphate buffer saline (PBS) to remove cell debris,
supplemented with serum-free medium, treated with 20, 40 or 80 µM
of luteolin, and incubated for 24 h at 37°C. The cells migrated
into the wound surface and the average distance of migrating cells
was determined under an IX81 Olympus inverted microscope (Olympus,
Tokyo, Japan) at 0 and 24 h.
Transwell migration assay
Transwell migration assays were performed, as
previously described (12).
Briefly, the cells were seeded into a 24-well Transwell plates
(Millipore, Bedford, MA, USA) in 10% FBS medium at a density of
1×105 cells/well. Following incubation for 24 h at 37°C,
the medium was replaced with serum-free medium and the cells were
treated with 20, 40 or 80 µM of luteolin or 300 nM Taxol for 24 h
at 37°C, while RPMI-1640 medium, containing 10% FBS was added to
the lower chamber. The NCI-H460 cells were treated with the drug
and cultured at 37°C for a further 5 h. The non-adherent cells were
removed by washing with PBS, and the adherent cells were fixed in
ethanol. Following staining with 0.1% crystal violet (Tokyo
Chemical Industry, Tokyo, Japan), images were captured by
microscopy (IX81; Olympus, Tokyo, Japan).
Western blot analysis
The NCI-H460 cells were seeded into a 6-well plate
at 3×105 cells/well and were incubated with 20, 40 or 80
µM luteolin or 300 nM Taxol for 24 h, washed once with PBS and the
cell lysates were prepared in radioimmunoprecipitation lysis
buffer, containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton
X-100 and 1 mM PMSF. The protein concentrations were determined
using a bicinchoninic acid protein assay kit (Beyotime Institute of
Biotechnology, Shanghai, China). The proteins (30 µg) were
resuspended in sample buffer containing 2% SDS, 2%
β-mercaptoethanol, 50 mmol/l Tris-HCl (pH 6.8), 10% glycerol and
0.05% bromophenol blue. The proteins were separated on an
SDS-polyacrylamide gel and transferred onto polyvinylidene fluoride
membranes (Millipore). Following blocking of the membrane with
Tris-buffered saline-0.1% Tween-20 (TBST), containing 2.5% bovine
serum albumin, for 1 h at room temperature, the membrane was washed
twice with TBST and incubated with primary antibodies overnight at
4°C. The primary antibodies were as follows: Rabbit anti-human
anti-Sirt1 (1:1,000; cat. no. 2496), rabbit anti-human anti-Bad
(1:1,000; cat. no. 4366), rabbit anti-human anti-Bcl-2 (1:1,000;
cat. no. 2827), rabbit anti-human anti-Bax (1:1,000; cat. no.
5023), rabbit anti-human anti-caspase-3 (1:1,000; cat. no. 9665)
and rabbit anti-human anti-cleaved caspase-3 (1:800). β-actin
(1:1,000; cat. no. 8456) was used as an internal loading control.
All antibodies were purchased from Cell Sigaling Technology, Inc.
(Danvers, MA, USA). Following incubation with the primary
antibodies, the membranes were washed three times for 10 mins each
with TBST and were subsequently incubated for 1 h at room
temperature with goat anti-rabbit immunoglobulin G horseradish
peroxidase-conjugated secondary antibodies (1:5,000; cat. no. 7074;
Cell Signaling Technologies, Inc.). Following washing three times
for 10 mins each in TBST, the bands were detected using enhanced
chemiluminescence reagent (Millipore). The band intensities were
quantified using a ChemiDoc™ MP system (io-Rad Laboratories, Inc.,
Hercules, CA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The total RNA from the NCI-H460 cells was extracted
using TRIzol reagent (Invitrogen Life Technologies, Grand Island,
NY, USA), according to the manufacturer's instructions. The RNA
concentration was determined using ultraviolet spectrophotometry
(NanoDrop2000; Thermo Fisher Scientific, Waltham, MA, USA). cDNA
was reverse transcribed from 1 µg total RNA using ReverTra Ace-α™
kit (Toyobo Co., Ltd., Osaka, Japan). A total of 2 µl template cDNA
was used for amplification. RT-qPCR was performed using Thunderbird
SYBR Master mix (Toyobo, Osaka, Japan). The primer sequences were
as follows: Sirt1, forward 5′-AAGGAGCAAGGTCGCTTACAGA-3′ and reverse
5′-CAAATGGCTTTCAGATAGTCAGGTC-3′ and GAPDH, forward
5′-GATCCCGCTAACATCAAATG-3′ and reverse 5′-GAGGGAGTTGTCATATTTCTC-3′,
and were all synthesized by GeneScript (Nanjing, China). RT-qPCR
was performed on a Step One plus system (ABI, Carlsbad, CA, USA),
using the following cycles: 95°C for 5 min and 40 cycles of 95°C
for 15 sec, 60°C for 30 sec and 72°C for 30 sec. The expression of
GAPDH was determined as an internal control. The 2−ΔΔCT
value was calculated for every sample and the mRNA expression
levels were determined using the 2−ΔΔCT method (13), normalized against GAPDH.
Annexin V/propidum iodide (PI) flow
cytometric analysis
Annexin V-fluorescein isothiocyanate (FITC)/PI
double staining was performed to quantitatively determine the
percentage of cells undergoing apoptosis. Briefly, the NCI-H460
cells were seeded into a 6-well plates at 2×105
cells/well and at 70–80% confluence, the cells were treated with
20, 40 or 80 µM luteolin or 300 nM Taxol at 37°C. Following
treatment for 48 h at 37°C, the cellular monolayer was released
using trypsin without EDTA (Gibco Life Technologies, Carlsbad, CA,
USA). The cells were resuspended and incubated with annexin V-FITC
(KeyGen Biotech Co., Ltd., Nanjing, China) for 15 min at room
temperature, followed by PI staining. The cells were analyzed using
a flow cytometer (Becton Dickinson, Mountain View, CA, USA).
Annexin V-FITC and PI double-negative cells were defined as normal
cells, whereas annexin V-FITC-positive and PI-negative cells were
defined as early apoptotic cells and annexin V-FITC and PI
double-positive cells were defined as late apoptotic and necrotic
cells. The annexin V-FITC-PI binding assay was determined at least
three times. CellQuest software (BD Biosciences San Jose, CA, USA)
was used to calculate the percentage distribution of normal, early
apoptotic, late apoptotic and necrotic cells.
Cell cycle analysis
To assess the cell cycle distribution,
2×104 cells/well were seeded into 6-well plates, treated
with 20, 40 or 80 µl luteolin and incubated for 24 h prior to
analysis. The cells were fixed with 70% ethanol at 4°C overnight
and were subsequently incubated with 100 µg/ml RNase A, and stained
with 50 µg/ml PI at room temperature. The DNA content was
determined using flow cytometry on a BD FACS Calibur flow cytometer
(BD, Franklin Lakes, NJ, USA). The distribution of the cell cycles
were analyzed using the ModFit LT software for Mac.
Lentiviral vectors
A Sirt1 short hairpin (sh)RNA expression vector was
constructed using pLentilHI, a lentiviral plasmid. The Sirt1 shRNA
oligonucleotides (sense 5′-GATCCGGAT GAA
AGTGAGATTGAATCAAGAGTTCAATCTCACTTT CATCCTTTTTTC-3′ and antisense
5′-TCGAGAAAAAAG GATGAAAGTGAGATTGAACTCTTGATTCAATCTCACT TTCATCCG-3′),
corresponding to the 2,055–2,074 sites of the porcine Sirt1 mRNA
(GenBank no. EU030283), were annealed and cloned into the pLentiHI
at the BamHI and XhoI sites (8).
Lentiviral infection
The pLentiHI-Sirt1 shRNA transferred plasmid (3.3
µg), 2 µg Δ8.9 packaging plasmid and 3 µg VSV-G envelope protein
plasmid were cotransfected into the HEK293T packaging cells
(2×105 cells/well) using Lipofectamine 2000 (Invitrogen
Life Technologies), according to the manufacturer's instructions.
Similarly, the FG30-Sirt1 or FG30 packaging plasmid and envelope
protein plasmid (Takara, Shiga, Japan) were contransfected into the
HEK293T packaging cells. Following transfection (48 h) at 37°C, the
supernatant, containing viral particles, was collected and passed
through a 0.45 µm filter (Merck Millipore, Darmstadt, Germany) to
remove cellular debris. Porcine preadipocytes were seeded at a
density of 1×105 cells/well and cultured in DMEM/F12
medium, containing 10% FBS at 37°C. At 70–80% confluence, the viral
suspension of Sirt1 shRNA and the scambled sequences, containing 6
mg/ml polybrene were added. Following infection for a further 48 h
at 37°C, the cells were harvested for analysis.
Statistical analysis
SPSS version 11.0 (SPSS, Inc., Chicago, IL, USA) for
Windows was used for all statistical analyses. The data obtained
from the different experiments are expressed as the mean ± standard
error of the mean, from at least three independent experiments and
were analyzed using Student-Newman-Keuls test. P<0.05 was
considered to indicate a statistically significant difference.
Results
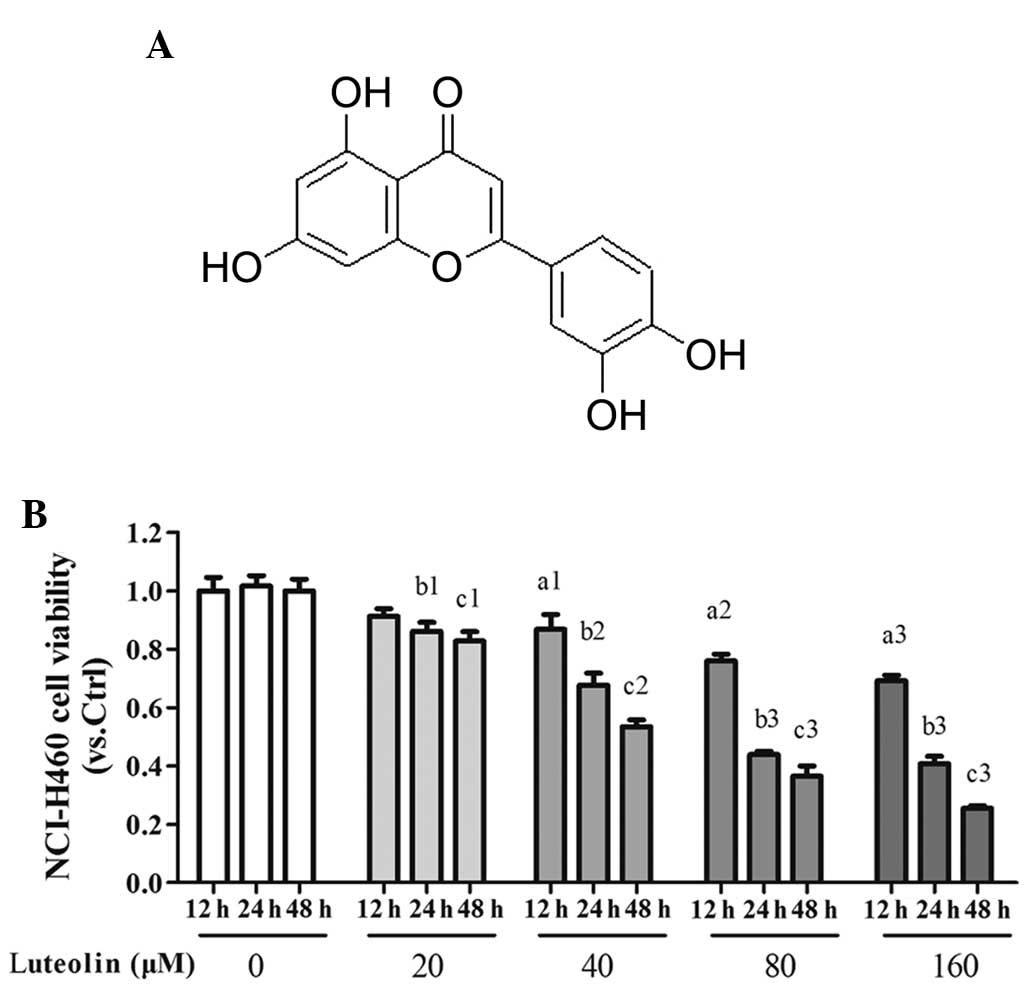
Antiproliferative effect of luteolin on
NCI-H460 cells in vitro
The aim of the initial experiments was to
investigate whether luteolin affected the viability of the NCI-H460
cell line in vitro. The range of concentrations assessed was
between 20 and 160 µM. As shown in Fig. 1, treatment of the NCI-H460 cells
for 24 h resulted in a concentration-dependent inhibition of the
mitochondrial oxidative metabolism, determined using an MTT assay.
The data revealed the inhibitor effect of luteolin on the viability
of the NCI-H460 cells in a concentration-dependent manner. In the
subsequent experiments, the NCI-H460 cells were treated for 24 h
with three concentrations, 20 40 and 80 µM, which caused a
reduction in cell viability by 86.06±9.23, 53.48±5.56 and
43.83±3.24%, respectively, in order to determine levels of
apoptosis of the NCI-H460 cells.
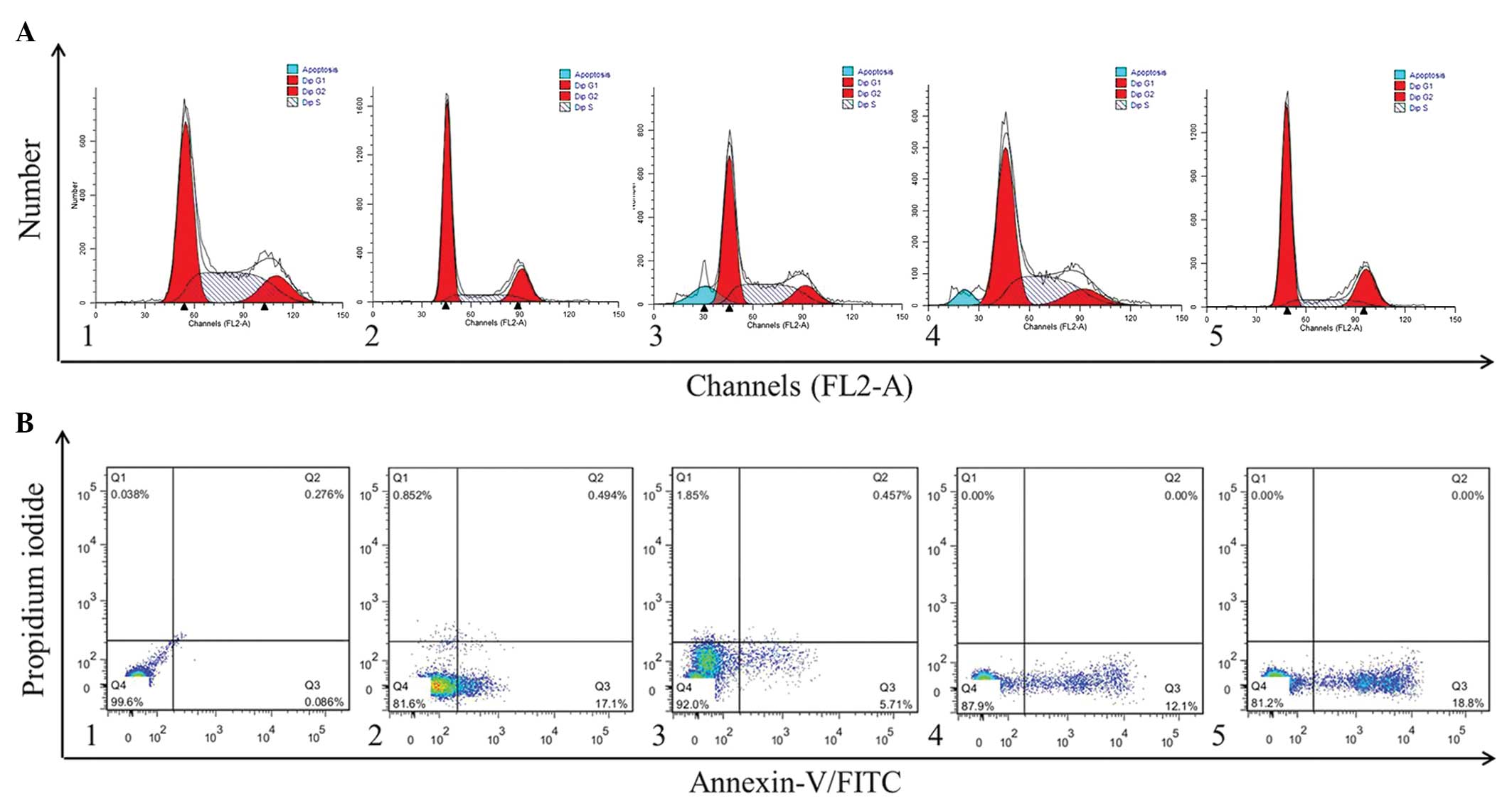
Effect of luteolin on the cell cycle and
apoptosis of NCI-H460 cells
The cell cycle distribution of the NCI-H460 cells
was analyzed using flow cytometry. The effect of luteolin treatment
for 24 h on cell cycle phase distribution is shown in Fig. 2A. A sub-G1 apoptotic peak was
observed, which is usually regarded as one of the characteristics
of apoptosis. Compared with the control cells, luteolin caused an
accumulation of cells in the S phase (Fig. 2A). In addition, the apoptotic
fraction was markedly increased following the addition of luteolin.
These results demonstrated that the reduction observed in the
proliferation of NCI-H460 cells mediated by luteolin was associated
with cell cycle arrest in the S phase.
Subsequent investigation of the effect of luteolin
on the apoptotic response of NCI-H460 cells, determined using
annexin V-FITC/PI staining (Fig.
2B), revealed that the apoptotic ratio increased in the
luteolin-treated NCI-H460 cells, compared with the cells in the
control group. These results demonstrated that luteolin suppressed
the proliferation of the NCI-H460 cells by inducing apoptosis.
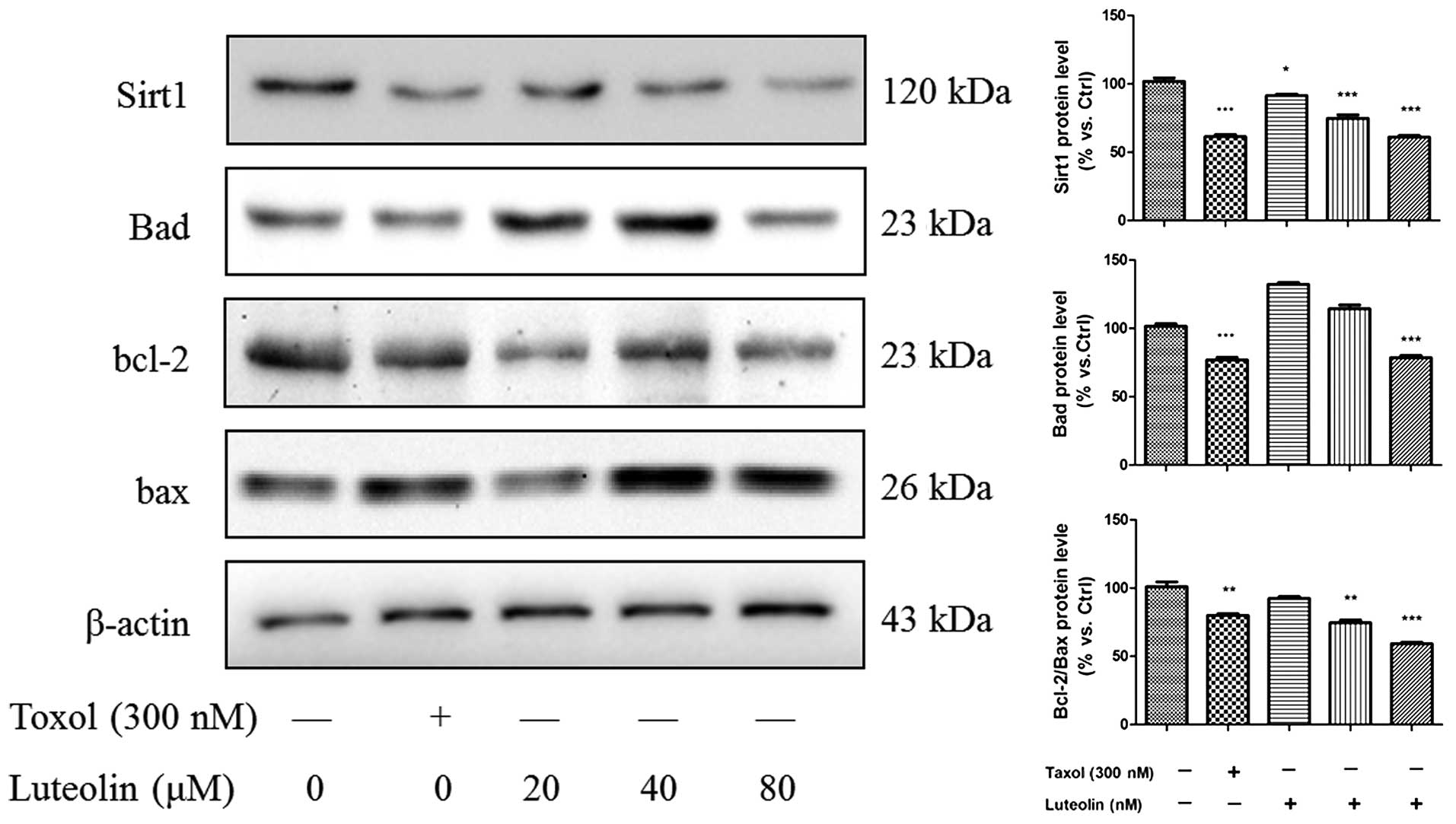
Effect of luteolin on
apoptosis-associated proteins
To confirm the effect of luteolin on apoptotic cell
death, NCI-H460 lysate was extracted from the groups treated with
different concentrations of the drug 24 h, and various
apoptosis-associated proteins were analyzed using western blotting
(Fig. 3). Luteolin increased the
protein expression levels of apoptotic regulatory proteins,
including the Bax/Bcl-2 ratio, in a concentration-dependent manner,
however, only 80 µM luteolin inhibited the expression of Bad.
Additionally, the expression of Sirt1 was analyzed in the presence
of various concentration of luteolin. The results demonstrated that
luteolin also decreased the expression of Sirt1 in the NCI-H460
cell line in a concentration-dependent manner.
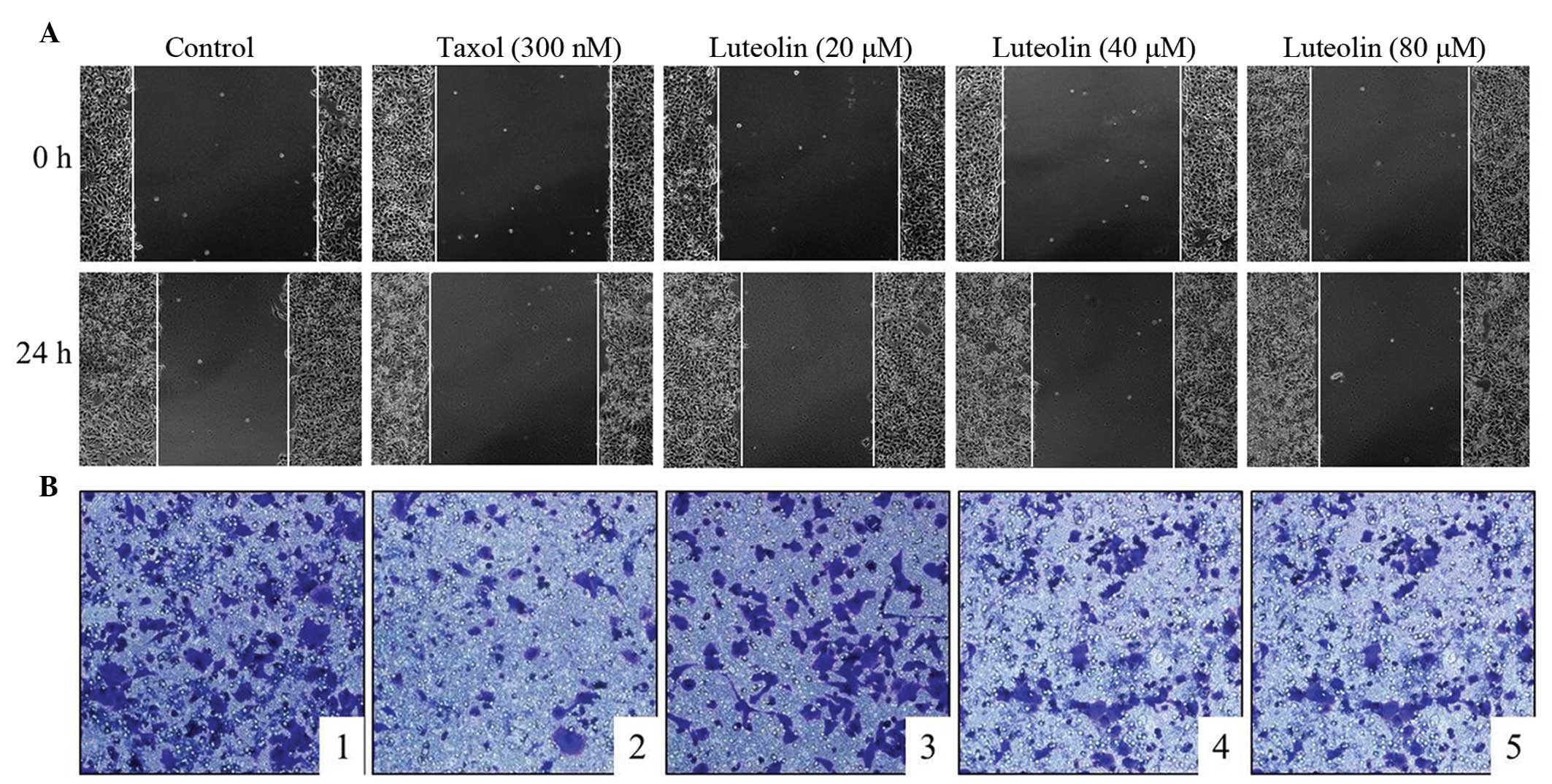
Effect of luteolin on NCI-H460 cell
migration
Cell migration is a hallmark of tumorgenesis and
metastasis (14). It is relevant
for angiogenesis to ensure tumor nutrition and for the formation of
metastases, in which tumor cells leave the primary tumor site and
spread to other tissues (15).
Therefore, anticancer agents are required not only inhibit tumor
cell growth, but also to prevent their metastases. As shown in
Fig. 4, the NCI-H460 cell lines
were treated with different concentrations of luteolin for 24 h and
cell migration was assessed using a wound healing assay (Fig. 4A) and Transwell assay (Fig. 4B). The results demonstrated that
luteolin exerted an inhibitory effect on the migration of the
NCI-H460 cells.
Sirt1 knockdown decreases the viability
and induces the apoptosis of NCI-H460
As the results suggested the potential role of Sirt1
in the regulation of NCI-H460 cell apoptosis, the present study
hypothesized that the sensitivity to camptothecin may be increased
by knockdown of Sirt1 using a lentivirus. The protein expression
levels of caspase-3, Bad, Bax and Bcl-2 in the Sirt1-knockdown
NCI-H460 cells were increased, compared with the empty vector and
scrambled shRNA. As shown in Fig.
5A, Sirt1 shRNA significantly decreased the protein expression
of Sirt1 in the NCI-H460 cells compared with the empty vector and
scrambled shRNA. Following knockdown of Sirt1, the activation of
caspase-3 in the NCI-H460 cells transfected with Sirt1 shRNA
increased significantly, and the expression levels of Bad and
Bcl-2/Bax were decreased, compared with the empty vector and
scrambled shRNA groups (Fig. 5B).
These findings suggested that Sirt1 regulated the apoptotic
response of NCI-H460 cells.
Discussion
The present study demonstrated that lueolin induced
the apoptosis of NCI-H460 cells and downregulated the expression of
Sirt1. It also demonstrated that the silencing of Sirt1 induced the
apoptosis of NCI-H460 cells. Luteolin had an anticancer effect on
NCI-H460 cells by affecting Sirt1-mediated apoptosis.
NCI-460, a human large cell lung cancer cell, is a
subtype of non-small cell lung carcinoma (NSCLC), which is the most
frequent type of lung cancer (16,17).
Sirt1, a nicotinamide adenosine dinucleotide-dependent histone
deacetylase, regulates the transcriptional activity of NF-κB
(18). The role of Sirt1 and
cortactin in unfavorable NSCLC characteristics and the progression
of ADC has been described previously (10), and its function in the induction of
cell apoptosis through the caspase-3 pathway in porcine
preadipocytes has been demonstrated (8). However, whether Sirt1 is important in
NSCLC remains to be elucidated.
Bax is a pro-apoptotic Bcl-2 family protein, which
resides in the cytosol and translocates to the mitochondria upon
the induction of apoptosis (19).
Caspase-3 is a key terminal-regulated apoptosis protein involved in
cellular apoptosis pathways. The present study demonstrated that
Sirt1 knockdown increased the activation of caspase-3 and decreased
the protein expression levels of Bad and Bcl-2/Bax, which induced
the apoptosis of the NCI-H460 cells (6,20).
Luteolin is a flavonoid, which has been observed to
exert anticancer activities, including the induction of apoptosis
and cell cycle arrest, and the inhibition of metastasis and
angiogenesis, in several cancer cell lines, including lung cancer
cells (5,16,21,22).
However, its effect on NCI-H460 cells remains to be fully
elucidated. Therefore, the present study investigated the
anticancer activity of luteolin on NCI-H460 cells. Luteolin
demonstrated inhibition of NCI-H460 cell viability in a
concentration-dependent manner, demonstrated using an MTT assay,
and caused alterations in the cell cycle and induced apoptosis,
determined using flow cytometry. Western blot analysis demonstrated
that luteolin inhibited the protein expression level of Bad and the
Bcl-2/Bax ratio, which can indicate the cell survival or apoptosis.
In addition, luteolin inhibited NCI-H460 cell migration in a
dose-dependent manner. These data indicated that luteolin may be a
novel and effective anticancer agent and, at 80 µM, has an effect
equal to that of 300 nM Taxol.
Notably, luteolin also inhibited the protein
expression levels of Sirt1 in the NCI-H460 cell line. Therefore, it
was suggested that the anticancer effect of luteolin on NCI-H460
cells was induced by Sirt1-mediated activation of the caspase-3
pathway. However, whether this effect is also induced by p53 and
c-JUN requires further investigation (9,23,24).
Acknowledgments
This study was supported by The National Key
Clinical Specialist Construction project of China (2011).
References
|
1
|
Szyszka-Barth K, Ramlau K,
Goździk-Spychalska J, et al: Actual status of therapeutic
vaccination in non-small cell lung cancer. Contemp Oncol (Pozn).
18:77–84. 2014.
|
|
2
|
Lin Y, Shi R, Wang X and Shen HM:
Luteolin, a flavonoid with potentials for cancer prevention and
therapy. Curr Cancer Drug Targets. 8:634–646. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lin Y, Shi R, Wang X and Shen HM:
Luteolin, a flavonoid with potential for cancer prevention and
therapy. Curr Cancer Drug Targets. 8:634–646. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Asgarpanah J and Kazemivash N:
Phytochemistry, pharmacology and medicinal properties of Carthamus
tinctorius L. Chin J Integr Med. 19:153–159. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cai X, Ye T, Liu C, et al: Luteolin
induced G2 phase cell cycle arrest and apoptosis on non-small cell
lung cancer cells. Toxicol In Vitro. 25:1385–1391. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Heltweg B, Gatbonton T, Schuler AD, et al:
Antitumor activity of a small-molecule inhibitor of human silent
information regulator 2 enzymes. Cancer Res. 66:4368–4377. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen HC, Jeng YM, Yuan RH, Hsu HC and Chen
YL: SIRT1 promotes tumorigenesis and resistance to chemotherapy in
hepatocellular carcinoma and its expression predicts poor
prognosis. Ann Surg Oncol. 19:2011–2019. 2012. View Article : Google Scholar
|
|
8
|
Pang WJ, Xiong Y, Wang Y, Tong Q and Yang
GS: Sirt1 attenuates camptothecin-induced apoptosis through
caspase-3 pathway in porcine preadipocytes. Exp Cell Res.
319:670–683. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yi J and Luo J: SIRT1 and p53, effect on
cancer, senescence and beyond. Biochim Biophys Acta. 1804:1684–9.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gao Z and Ye J: Inhibition of
transcriptional activity of c-JUN by SIRT1. Biochem Biophys Res
Commun. 376:793–796. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shi H, Wang J, Dong F, Wang X, Li H and
Hou Y: The effect of proteoglycans inhibited by RNA interference on
metastatic characters of human salivary adenoid cystic carcinoma.
BMC Cancer. 9:4562009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Daoud A, Song J, Xiao F and Shang J:
B-9-3, a novel beta-carboline derivative exhibits anti-cancer
activity via induction of apoptosis and inhibition of cell
migration in vitro. Eur J Pharmacol. 724:219–230. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method[J]. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
14
|
Yamaguchi H, Wyckoff J and Condeelis J:
Cell migration in tumors. Curr Opin Cell Biol. 17:559–564. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sahai A, Mallina R, Dowson C, Larner T and
Khan MS: Evolution of transdermal oxybutynin in the treatment of
overactive bladder. Int J Clin Pract. 62:167–170. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Park SH, Park HS, Lee JH, et al: Induction
of endoplasmic reticulum stress-mediated apoptosis and
non-canonical autophagy by luteolin in NCI-H460 lung carcinoma
cells. Food Chem Toxicol. 56:100–109. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kollipara PS, Jeong HS, Han SB and Hong
JT: (E)-2,4-bis (p-hydroxyphenyl)-2-butenal has an
antiproliferative effect on NSCLC cells induced by p38
MAPK-mediated suppression of NF-kappaB and up-regulation of
TNFRSF10B (DR5). Br J Pharmacol. 168:1471–1484. 2013. View Article : Google Scholar :
|
|
18
|
Yeung F, Hoberg JE, Ramsey CS, et al:
Modulation of NF-κB-dependent transcription and cell survival by
the SIRT1 deacetylase. EMBO J. 23:2369–2380. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun Y, Sun D, Li F, et al: Downregulation
of Sirt1 by antisense oligonucleotides induces apoptosis and
enhances radiation sensitization in A549 lung cancer cells. Lung
Cancer. 58:21–29. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen MC, Chen CH, Liu YN, Wang HP, Pan SL
and Teng CM: TW01001, a novel piperazinedione compound, induces
mitotic arrest and autophagy in non-small cell lung cancer A549
cells. Cancer Lett. 336:370–378. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chian S, Thapa R, Chi Z, Wang XJ and Tang
X: Luteolin inhibits the Nrf2 signaling pathway and tumor growth in
vivo. Biochem Biophys Res Commun. 447:602–608. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bai L, Xu X, Wang Q, et al: A
superoxide-mediated mitogen-activated protein kinase phosphatase-1
degradation and c-Jun NH2-terminal kinase activation pathway for
luteolin-induced lung cancer cytotoxicity. Mol Pharmacol.
81:549–555. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Solomon JM, Pasupuleti R, Xu L, et al:
Inhibition of SIRT1 catalytic activity increases p53 acetylation
but does not alter cell survival following DNA damage. Mol Cell
Biol. 26:28–38. 2006. View Article : Google Scholar :
|
|
24
|
Liu X, Shao K and Sun T: SIRT1 regulates
the human alveolar epithelial A549 cell apoptosis induced by
Pseudomonas aeruginosa lipopolysaccharide. Cell Physiol Biochem.
31:92–101. 2013. View Article : Google Scholar : PubMed/NCBI
|