Introduction
Adenoid cystic carcinoma (ACC) is an aggressive type
of malignancy, which develops in major and minor salivary glands
and rarely in other sites. ACC accounts for ~10% of all neoplasia
of the salivary glands, 22% of all salivary gland malignancies, and
~1% of all head and neck malignancies (1–3). A
hallmark of ACC is its aggressive ability to invade and
metastasize. It is currently accepted that local disease requires
treatment by local radical excision and postoperative radiotherapy,
while chemotherapy may have a limited benefit for advanced ACC
(4,5). Tumor cells are characterized by high
proliferation rates and demand for oxygen and nutrients. However,
the abnormal structure and function of the vascular network leads
to inadequate blood flow and fails to satisfy tumor cell demand for
oxygen and nutrients, which contributes to the formation of a
hypoxic microenvironment (4). It
is widely accepted that hypoxia is intricately associated with
tumor aggressiveness, angiogenesis, increased rates of reoccurrence
and chemotherapy resistance (6).
Hypoxia-inducible factor-1 (HIF-1) is an important
heterodimeric transcription factor composed of a labile HIF-1α
subunit and a stable HIF-1β subunit (7). Under normoxic conditions, HIF-1α is
degraded proteasomally; whereas under hypoxic conditions, HIF-1α is
stabilized and moves to the nucleus where it freely forms a complex
with HIF-1β, which in turn binds to hypoxia response elements and
induces the transcription of several genes, including >60 genes
activated by HIF-1 (8,9). The expression of HIF-1α has been
detected in ordinary and transformed ACC, and occasionally in
normal salivary tissues adjacent to the tumor (10). Previously, HIF-2α, one of the three
homologues of the HIFα subunit, has also been observed in ACC
tissues, and correlated with invasion and metastasis (11). On the basis of these findings, HIFs
may be important in the aggressive behavior of ACC.
Autophagy is an important intracellular process
involved in the degradation and recycling of cytosolic material,
which is essential for the maintenance of cellular biosynthesis
(12). Accumulating evidence
supports a controversial role for autophagy in cancer, as autophagy
enables tumor cells to tolerate adverse stress conditions,
including hypoxia, and autophagy may protect cells through damage
mitigation, which may limit tumorigenesis (13). Our previous study investigated 79
patients with head and neck ACC, and observed the expression levels
of autophagy-associated protein 1 light chain 3 (LC3) and Beclin 1.
The results indicated that LC3 and Beclin 1 may be important in the
tumorigenesis of ACC (14).
Furthermore, inhibition of autophagy enhanced chemotherapy efficacy
in ACC (15), and these results
were concordant with those of and Ma et al (16). Therefore, further studies on
autophagy and a better understanding of the role of autophagy in
ACC is essential for future developments in its treatment
Previous studies have uncovered several associations
between hypoxia and the induction of autophagy (12,13).
However, the molecular mechanism underlying hypoxia-induced
autophagy in ACC remains to be elucidated. Considering the
correlation between hypoxia and tumor invasion in ACC (11), whether hypoxia-induced autophagy is
associated with tumor invasion in ACC also remains to be
elucidated. In the present study, ACC-M cell lines were treated
with the hypoxia mimetic cobalt chloride (CoCl2) to
investigate the levels of hypoxia mimetic-induced autophagy in
salivary ACC cells and its effects on tumor invasion.
Materials and methods
Chemical reagents and antibodies
CoCl2, chloroquine (an inhibitor of
autophagy) and antibodies targeting LC3B were purchased from
Sigma-Aldrich (St. Louis, MO, USA). Antibodies targeting HIF-1α and
B cell lymphoma 2 (Bcl-2)/adenovirus E1B 19K-interacting protein 3
(BNIP3) were purchased from Santa Cruz Biotechnology, Inc. (Dallas,
TX, USA). Antibodies targeting Beclin 1 and GAPDH were purchased
from Abcam (Cambridge, MA, USA).
Cell culture
The ACC-M cell lines were obtained from Professor
Wantao Chen (Department of Oral and Maxillofacial Surgery, Ninth
People's Hospital, College of Stomatology, Shanghai Jiao Tong
University, Shanghai, China), and were maintained in culture with
Dulbecco's modified Eagle's medium (Invitrogen Life Technologies,
Carlsbad, CA, USA) supplemented with 10% fetal bovine serum
(Invitrogen Life Technologies) in a humidified atmosphere
containing 5% CO2 at 37°C. To induce a hypoxic
environment, the cells were plated on a 35 mm dish and, after 24 h,
were cultured in medium supplemented with CoCl2 in an
atmosphere containing 5% CO2 at 37°C.
Cell viability assay
Cell viability assays were performed using an MTT
assay (Sigma-Aldrich). Briefly, the cells were plated into 96-well
plates at a density of 1×104 cells/well. Following
overnight incubation, the cells were treated with the indicated
concentrations of CoCl2 (50, 100, 150, 200, 250, 300,
500 and 1,000 µmol/l) for 24 h. A total of 20 µl MTT
solution (5 mg/ml) was subsequently added to each well and
incubated for 4 h at 37°C. Blotting nutrient solution and 150
µl dimethyl sulfoxide (DMSO; Sigma-Aldrich) were placed in
each well. Following agitation for 10 min, the absorption value of
each well was measured at 570 nm using an enzyme-linked immune
detector (Softmax PRO M2, Molecular Devices, Sunnyvale, CA, USA).
Cell viability was expressed as the percentage of viable cells
relative to the untreated cells. All experiments were performed in
triplicate.
Transmission electron microscopy
(TEM)
TEM was performed, as previously described (15). The ACC-M cells (1×106)
were incubated with DMSO (control) and 200 µM
CoCl2 for 24 h at 37°C. The cells were then fixed with
2% glutaraldehyde (Beijing Chemical Industry Group, Co., Ltd.,
Beijing, China) in 0.1 M Sorensen buffer (pH 7.3; Beijing Chemical
Industry Group, Co., Ltd.) for 1 h at 4°C, and post-fixed in 1%
osmium tetroxide (Beijing Chemical Industry Group, Co., Ltd.) in
0.1 M cacodylate buffer (Beijing Chemical Industry Group, Co.,
Ltd.) for 1 h at room temperature. The specimens were dehydrated
through a graded series of ethanol (30–90%), and embedded in Epon
(Beijing Chemical Industry Group, Co., Ltd.). Following staining
with 2% uranyl acetate (Beijing Chemical Industry Group, Co.,
Ltd.), thin sections (50–70 nm) were prepared with a UC7 microtome
(Leica, Wetzlar, Germany). were observed using a JEM-1200EX
Transmission Electron Microscope (JEOL, Ltd., Tokyo, Japan).
Immunofluorescence staining
A total of 2×105 ACC-M cells were grown
on coverslips in 6 cm dishes, allowed to attach by overnight
incubation at 37°C, and then cultured with DMSO (control) or 200
µM CoCl2 for 24 h. At the end of treatment, the
cells were washed with phosphate-buffered saline (PBS), fixed with
4% paraformaldehyde (Beijing ComWin Biotech Co., Ltd., Beijing,
China); for 20 min, washed twice with PBS for 5 min and
permeabilized with 0.5% Triton X-100 (Beijing ComWin Biotech Co.,
Ltd.) for 10 min. Following washing with PBS, the cells were
incubated with primary antibody at 4°C overnight. For LC3
localization detection, the cells were incubated with rabbit
anti-LC3B antibody (1:50 diluted in 5% non-fat milk, polyclonal,
cat. no. L8918); fluorescein isothiocyanate (FITC)-conjugated goat
anti-rabbit immunoglobulin (Ig) G (1:50 diluted in 5% non-fat milk,
cat. no. cw0114) (both from Beijing ComWin Biotech Co., Ltd.,
Beijing, China) for 1 h at 4°C. The nuclei were then counterstained
with DAPI (Beijing ComWin Biotech Co., Ltd.); for 7 min, and
observed under a fluorescence microscope ((TE2000; Nikon
Corporation, Tokyo, Japan). In order to further identify the
hypoxia-induced autophagy pathway, double immunofluorescence
labeling was performed by simultaneous incubation of mouse
anti-BNIP3 (1:100 diluted in 5% non-fat milk, monoclonal, cat. no.
sc56167); rabbit anti-HIF-1α (1:100 diluted in 5% non-fat milk,
polyclonal, cat. no. sc10790); tetramethylrhodamine-conjugated goat
anti-mouse IgG (1:50 diluted in 5% non-fat milk, cat. no. cw0167)
(both from Beijing ComWin Biotech Co., Ltd, Beijing, China) for 1 h
at 37°C. The subsequent staining procedure was performed, as
described for the LC3B-positive cells.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells using TRIzol
reagent (Takara Biotechnology, Co., Ltd., Dalian, China), according
to the manufacturer's instructions. Reverse transcription of 1
µg total RNA was performed with the PrimeScript™ RT reagent
kit (Takara Biotechnology, Co., Ltd.) with gDNA Eraser (Takara
Biotechnology, Co., Ltd.), according to the manufacturer's
instructions. The total 20 µl reacting solution included 10
µl SYBR® Green mix (Takara Biotechnology, Co.,
Ltd.), 0.8 µl PCR forward primer, 0.8 µl PCR reverse
primer, 0.4 µl ROX reference dye, 2 µl cDNA and 6
µl dH2O. The ACC-M cells were detached and
homogenized using TRIzol® (Takara Biotechnology Co.,
Ltd.) and total RNA was extracted. RT-qPCR was performed using a
Roche Light Cycler 480 (Roche Diagnostics GmbH, Mannheim, Germany)
in a total volume of 20 µl reacting solution. The following
gene-specific primers were used: HIF-1α forward,
5′-CATCAGTTGCCACTTCCACATAA-3′ and reverse,
5′-GAGAACCATAACAAAACCATCCAAG-3′; BNIP3 forward,
5′-GCTTCTGAAACAGATACCCATAGCA-3′ and reverse,
5′-CGACTTGACCAATCCCATATCC-3′; LC3B forward,
5′-AAACGCATTTGCCATCACA-3′ and reverse,
5′-GGACCTTCAGCAGTTTACAGTCAG-3′; and β-actin forward,
5′-TGGCACCCAGCACAATGAA-3′ and reverse,
5′-CTAAGTCATAGTCCGCCTAGAAGCA-3′ (Takara Biotechnology Co., Ltd.).
The cycling conditions were identical for all primers.
SYBR® Green Mix (Takara Biotechnology Co., Ltd.) was
used to determine the abundance of mRNA, and the mRNA levels were
calculated relative to those of β-actin. The reaction conditions
were as follows: Stage 1, one cycle as 95°C for 30 sec; stage 2, 40
cycles at 95°C for 5 sec, and 60°C for 34 sec; followed by a
dissociation stage (95°C for 15 sec; 60°C for 1 min; 95°C for 15
sec). The comparative threshold cycle (2−ΔΔCT) method
was used to quantify the mRNA expression levels of these genes.
Western blot analysis
To extract the total protein of the cells, the
treated cells were washed three times with ice-cold PBS, and then
lysed in lysis buffer (RAPA:phenylmethylsulfonyl fluoride, 100:1;
Shennong Bocai Biotechnology Co., Ltd., Shanghai, China). A
bicinchoninic acid protein assay kit (Shennong Bocai Biotechnology
Co., Ltd.) was used to quantify the total protein content,
expressed in mg/ml. Samples containing equal quantities of protein
were resolved using 10–15% SDS-PAGE gels (Shennong Bocai
Biotechnology Co., Ltd.) and transferred onto polyvinylidene
fluoride membranes (Shennong Bocai Biotechnology Co., Ltd.). The
membranes were then incubated with primary antibodies targeting
HIF-1α (1:750), BNIP3 (1:1,000), LC3B (1:1,000) and Beclin 1
(1:1,000) overnight. The membranes were subsequently incubated with
peroxidase-conjugated secondary antibodies for 60 min. Protein
bands incubated with enhanced chemiluminescence reagents (Shennong
Bocai Biotechnology Co., Ltd.) were visualized using an Alpha
Imager 2200 system (Alpha Innotech Corporation, San Leandro, CA,
USA). To ensure equal protein loading, each membrane was stripped
and reprobed with anti-GAPDH antibody (1:1,000). GAPDH was used as
a control for protein level quantification. The band density was
quantified by the Quantity One image processing program, version
4.62 (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Cell invasion assay
An invasion assay was performed using a BD BioCoat
Growth Factor Reduced Matrigel Invasion Chamber (BD Biosciences,
San Jose, CA, USA), according to the manufacturer's instructions.
An equivalent number of cells (5×104/well) was
resuspended in serum-free medium, with or without CoCl2
(200 µM), and plated in the upper insert. To further examine
the effects of autophagy inhibition on invasion under hypoxia,
another group of cells were incubated in the presence or absence of
chloroquine (CQ; 10 and 20 µM; Sigma-Aldrich) with 200
µM CoCl2. The insert was then placed inside a
24-well plate. The cells were incubated for 24 h at 37°C, following
which the upper surface of the membrane in each insert was gently
wiped with a cotton swab to remove all non-invading cells. The
cells on the under surface were fixed with 4% paraformaldehyde for
15 min. The insert was subsequently placed in 0.1% crystal violet
(Beijing Chemical Industry Group, Co., Ltd.) to stain the cells.
The numbers of cells in seven randomly-selected fields of each
filter were estimated by manual counting, and each experiment was
performed independently at least three times. Leica-DM4000B
microscope.
Statistical analysis
The data are expressed as the mean ± standard error
of the mean of at least three independent experiments. Statistical
comparisons between groups were performed using two-tailed
Student's t-test. Statistical analysis was performed using
SPSS 15.0 (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered
to indicate a statistically significant difference.
Results
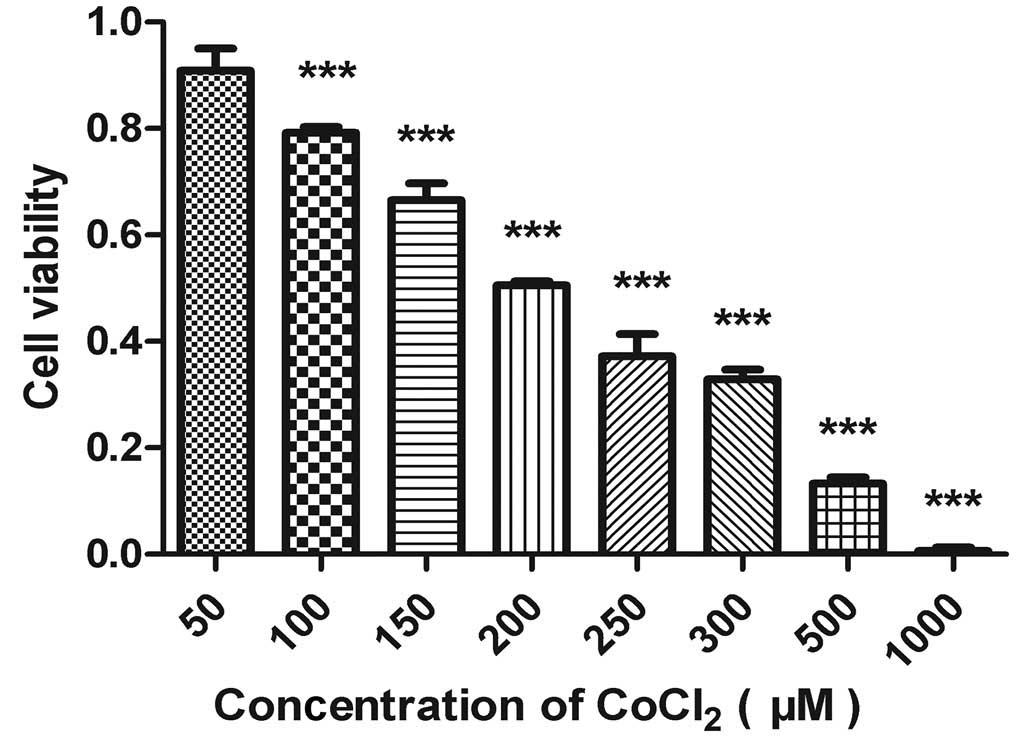
Cell viability assay
In the present study, ACC-M cells were treated with
hypoxia mimetic CoCl2. An MTT assay was used to quantify
the viability of the ACC-M cells treated with CoCl2. As
shown in Fig. 1, CoCl2
markedly reduced the viability of the ACC-M cells in a clear
dose-dependent manner. According to the experimental data, the 50%
inhibitory concentration of CoCl2 in the culture system
was ~200 µmol/l.
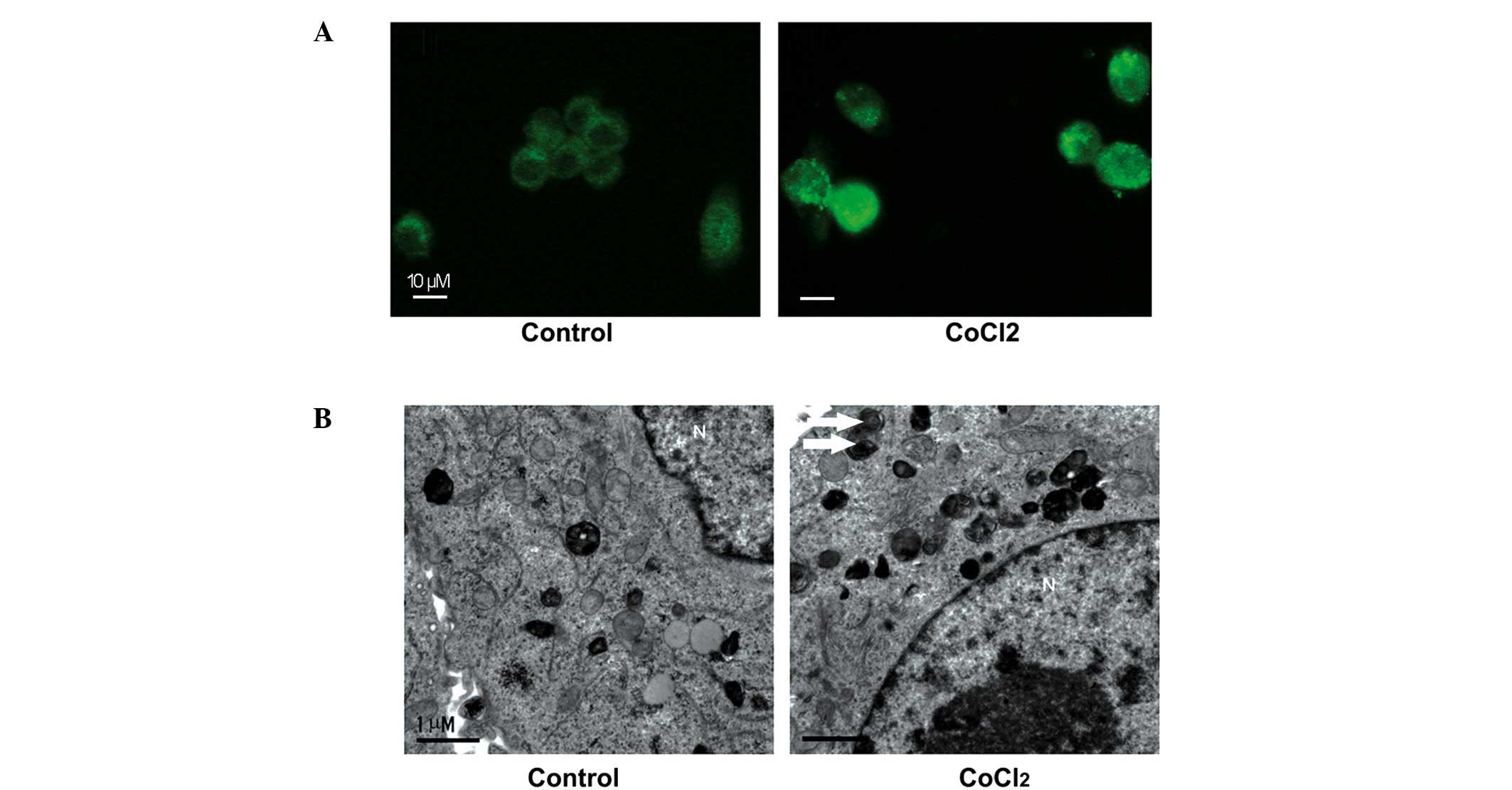
Autophagosome formation in ACC-M cells in
response to CoCl2
Our previous study reported that
autophagy-associated protein expression was observed in ACC tissue
samples (14). In order to examine
whether hypoxia induced autophagy in the ACC-M cells, the present
study used morphological analyses. LC3-II is conjugated with
phosphatidylethanolamine and is present on isolated membranes and
autophagosomes. Although LC3 has several homologs in mammals, LC3B
is most commonly used for autophagy assays (17). Therefore, immunostaining with LC3B
was used in the present study to observe the formation of
autophagosomes. Increased numbers of cytoplasmic puncta tethered
with LC3B were present in the CoCl2-treated ACC-M cells,
compared with the untreated cells (Fig. 2A). These results suggested the
formation of autophagosomes in the ACC-M cells. TEM was used for
further confirmation. Numerous autophagosome-like structures
consisting of double membranes were observed in the
CoCl2-treated ACC-M cells, compared with untreated
cells, which was indicative of autophagosome formation (Fig. 2B). In conclusion, these results
indicated that the CoCl2 mimetic hypoxia induced the
formation of autopha-gosomes in the ACC-M cells.
Involvement of the HIF-1α/BNIP3 signaling
pathway in CoCl2-induced autophagy in ACC-M cells
Increasing evidence has demonstrated that the
HIF-1-dependent signaling pathway is important in hypoxia-induced
autophagy (18). In addition,
BNIP3, a downstream gene regulated by HIF-1, has been demonstrated
to be essential for the HIF-1-dependent signaling pathway in
several cell lines (19,20). The present study demonstrated the
involvement of the HIF-1α/BNIP3 signaling pathway in
CoCl2-induced autophagy in ACC. As shown in Fig. 3A, treatment of the ACC-M cells with
200 µmol/l CoCl2 resulted in a marked increase in
the protein expression levels of HIF-1α and BNIP3 at different
time-points. Double immunofluorescence labeling of HIF-1α and BNIP3
confirmed the expression of HIF-1α in the nucleus, and expression
of BNIP3 in the cytoplasm (Fig.
3D), whereas fluorescence in the untreated cells was limited
(data not shown). As shown in Fig.
3C, the mRNA expression levels of HIF-1α were not affected by
CoCl2 treatment; however, following 24 h of treatment
with CoCl2, a ~6-fold increase in the mRNA expression of
BNIP3 was observed (P<0.001). With regards to
autophagy-associated gene expression in ACC, a marked increase in
the expression of LC3B-II was observed, however, minimal change was
detected in the expression of LC3B-I (Fig. 3B). The mRNA expression levels of
LC3B were also significantly increased following 24 h of treatment
with CoCl2 (P<0.001; Fig. 3C). In addition, an increase in the
expression of Beclin 1 was detected using western blot analysis
(Fig. 3B). These results suggested
that the mimetic hypoxia induced autophagy in the ACC-M cells and
that the HIF-1α/BNIP3 signaling pathway is essential for
hypoxia-induced autophagy in ACC.
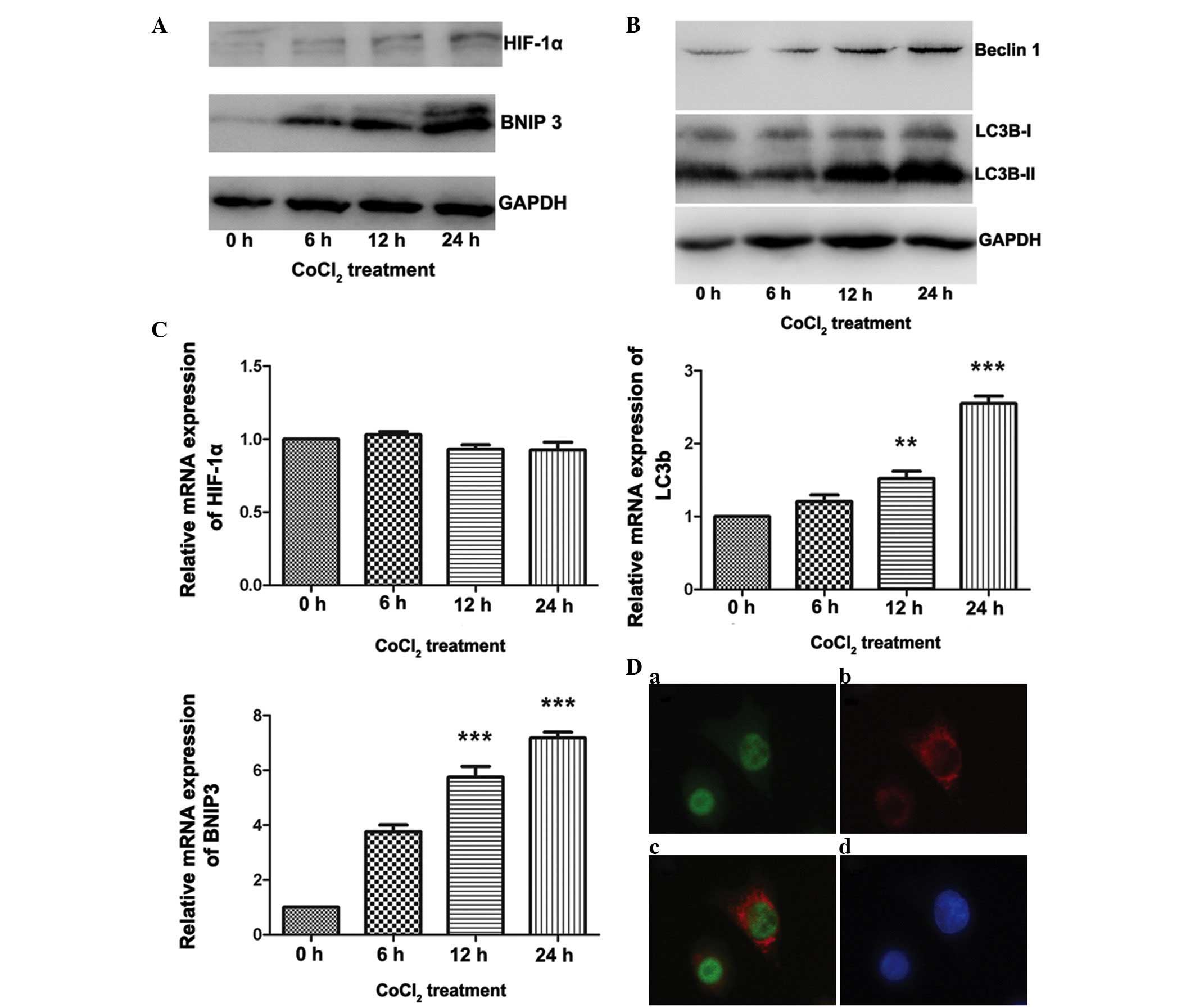 | Figure 3HIF-1α/BNIP3 pathway is activated in
ACC-M cells treated with CoCl2. The ACC-M cells were
cultured with 200 µM CoCl2 for the indicated
time-periods. Untreated cells (0 h group) served as a control. (A)
Variations in the protein expression levels of HIF-1α and BNIP3
were compared using western blotting, with GAPDH as an internal
control. (B) Expression levels of autophagy-associated proteins
were increased in the ACC-M cells treated with CoCl2.
The ACC-M cells were cultured with 200 µM CoCl2
for the indicated time-periods. Untreated cells t (0 h group)
served as a control. Variations in the protein expression levels of
Beclin 1 and LC3 were compared using western blotting, with GAPDH
as an internal control. (C) Variations in the mRNA expression
levels of HIF-1α, BNIP3 and Beclin1 were examined using reverse
transcription-quantitative polymerase chain reaction. The ACC-M
cells were cultured with 200 µM CoCl2 for the
indicated time-periods. Untreated cells (0 h group) served as a
control. *P<0.05, **P<0.01 and
***P<0.001, vs. control group. The data are presented
as the mean ± standard error of the mean of three independent
experiments with duplicates. (D) Double immunofluorescent staining
for HIF-1α and BNIP3 in the ACC-M cells treated with
CoCl2. (a) HIF-1α, (b) BNIP3, (c) merged signals, (d)
DAPI staining in the nucleus. The staining was observed by
fluorescence microscopy; magnification, ×400. HIF-1α,
hypoxia-inducible factor 1α; BNIP3, B cell lymphoma 2/adenovirus
E1B 19K-interacting protein 3; CoCl2, cobalt chloride;
ACC, adenoid cystic carcinoma. |
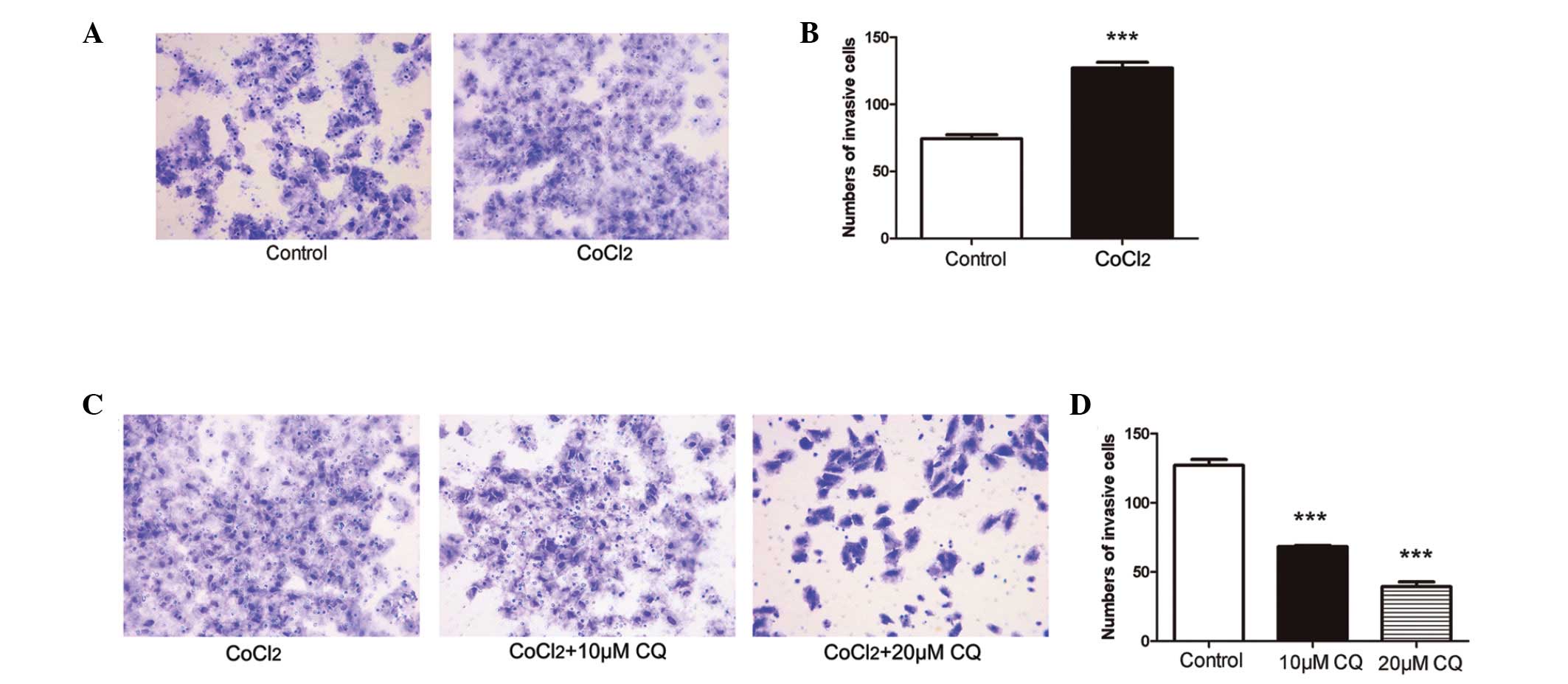
Inhibition of autophagy attenuates tumor
invasion induced by hypoxia in ACC-M cells
In the present study, a cell invasion assay was used
to determine the effect of mimetic hypoxia on tumor invasion.
Compared with the untreated cells, invasive cell numbers under
mimetic hypoxia were significantly increased (P<0.001; Fig. 4A and B). To further examine the
role of hypoxia-induced autophagy in tumor invasion in ACC, the
ACC-M cells were treated with or without CQ (10 µM and 20
µM) under mimetic hypoxia. The number of invading ACC-M
cells under mimetic hypoxia was markedly reduced 24 h after CQ
treatment (Fig. 4C and D), and
higher concentrations of CQ had an increased effect on the
inhibition of tumor invasion, compared with lower concentrations
(P<0.001).
Discussion
Our previous study reported that autophagy may be
important in the tumorigenesis of ACC (14). Considering the ubiquitous existence
of hypoxia in ACC (10,11), the present study investigated
whether hypoxia induces autophagy in ACC, and investigated its role
in tumor invasion. To the best of our knowledge, the present study
is the first to report the effects of CoCl2 on autophagy
in ACC. In the present study, mimetic hypoxia induced by
CoCl2 markedly upregulated the formation of
autophagosome, and the expression levels of autophagy-associated
genes. In addition, the results demonstrated that HIF-1α and its
target gene, BNIP3, are important in hypoxia-induced autophagy in
ACC. Exposure to CoCl2 increased the number of invading
cells, and inhibition of autophagy attenuated hypoxia-induced tumor
invasion in ACC-M cells.
Hypoxia develops in tumors due to incomplete
vascular supply and increasing demand for oxygen, compared with
normal tissues (4). Accumulating
evidence suggests that hypoxia exists in the majority of types of
head and neck cancer and is indicated as an important negative
factor (4). CoCl2 has
been widely used to mimic hypoxia, which can disturb the
degradation of HIF-1α and lead to its overexpression (21). Previous studies have demonstrated
that CoCl2 induces apop-tosis and autophagic cell death
in several cell types, including human periodontal ligament cells
(22) and neuroblastoma cells
(23), and increasing
CoCl2 doses affects cell viability in a dose-dependent
manner (24), which was also
demonstrated in in ACC-M cells in the present study.
As a transcriptional factor, HIF-1α moves to the
nucleus and forms a complex with HIF-1β, where it induces the
transcription of numerous genes, including BNIP3 (7–9).
BNIP3 is a BH3-only Bcl-2 family member regulated by HIF-1, and has
been identified as one of the most prominent hypoxia-responsive
genes (25). Under hypoxic
conditions, the expression levels of BNIP3 and BNIP3L are elevated
and contribute to hypoxia-induced cell death (25). Vengellur et al (26) demonstrated that the expression of
BNIP3 is HIF-1α-dependent, and CoCl2 induces their
expression in a time- and dose-dependent manner in mouse embryonic
fibroblasts (26). Concordant with
these studies, the protein and mRNA expression levels of BNIP3 were
elevated following CoCl2 treatment in a time-dependent
manner in the present study.
Autophagy is a lysosomal degradation pathway, which
involves the degradation and recycling of cytoplasm material.
Autophagy is characterized by at least four fundamental steps:
Induction; entrapment of organelles and cytoplasm in
double-membrane vesicles, termed autophagosomes; autophagosome
docking and fusion with the lysosome or vacuole; and autophagic
body degradation (27). Hypoxia is
able to rapidly induce autophagy in an HIF-1-dependent pathway. By
investigating HIF-knockdown cells, Bohensky et al (28) demonstrated that HIF-1 modulates the
induction of autophagic proteins by regulating the association
between Bcl-2 and Beclin 1. In a previous study on the functional
and physical interactions between Bcl-2 and Beclin 1, the BH3
domain was demonstrated to be involved in autopahgy (29,30).
Consequently, members of the BH3-only subfamily, including BNIP3,
are under further investigation. Bellot et al (19) demonstrated that the atypical BH3
domains of hypoxia-induced BNIP3/BNIP3L induce autophagy by
disrupting the Bcl-2/Beclin 1 complex without inducing cell death
(19). These findings suggest that
the HIF-1α/BNIP3 signaling pathway may be important in mimetic
hypoxia-induced autophagy in ACC. The results of the present study
demonstrated that exposure to CoCl2 resulted in
overexpression of HIF-1 and activation of BNIP3, as well as
upregulation of autophagosome formation and in the expression
levels of Beclin 1 and LC3-II. Therefore, the results of the
present study demonstrated that mimetic hypoxia by CoCl2
was able to induce autophagy via the HIF-1α/BNIP3 signaling pathway
in ACC.
By affecting the degradation of the basement
membrane and extracellular matrix (ECM), modulation of cell
adhesion molecules and cell migration, hypoxia is able to promote
tumor invasion and metastasis (31). In ACC-M cells in the present study,
exposure to mimetic hypoxia markedly increased tumor invasion. A
previous study demonstrated that hypoxia-induced autophagic stroma,
including fibroblasts and ECM, are important in tumor invasion and
metastasis via paracrine secretion (27).
Whether autophagy of tumor cells is involved in the
process of invasion and metastasis remains to be fully elucidated,
therefore, investigations to examine the effects of autophagy on
tumor invasion are required. Macintosh et al (32) investigated the role of autophagy in
tumor cell invasion using a three-dimensional (3D) organotypic
model. The results demonstrated that inhibition of autophagy by
shAtg12 failed to affect cell migration, but impaired tumor
invasion in the 3D organotypic model (32). These results are concordant with
the findings of the present study, which demonstrated that
inhibition of autophagy attenuated hypoxia-induced tumor invasion
in ACC-M cells. To further investigate the molecular mechanism
underlying the role of autophagy in tumor invasion, Li et al
(33) reported that autophagy
promoted hepatocellular carcinoma cell invasion through activation
of transforming growth factor β/Smad3 signaling during starvation.
Yamanaka-Tatematsu et al (34) demonstrated that the autophagy
induced by CoCl2 treatment increases tumor invasion
through supplementation of adenosine triphosphate (ATP). These
findings suggested that recruitment of ATP supplied by autophagy
may be vital to tumor invasion. These results are concordant with
those of a previous report that demonstrated that ATP induces the
release of matrix metal-loproteinase 9 to support invasion
(35). Indelicato et al
(36) reported that activation of
autophagy also inhibits tumor invasion via the induction of HIF-1α
degradation by autophagy. However, whether inhibition or activation
of autophagy attenuates tumor invasion remains to be elucidated. In
the present study, CQ was selected as an autophagic inhibitor. CQ
and hydroxyl-CQ are the only autophagic inhibitors used in clinical
trials at present (12). The
results of the present study indicated that combining traditional
treatment with autophagic target therapy may be efficient in
suppressing tumor invasion of ACC. However, identifying the
regulatory mechanism underlying autophagy in tumor invasion is
important in order for autophagy to be targeted in future
treatment.
In conclusion, the results of the present study
revealed that CoCl2 mimetic hypoxia induced autophagy in
ACC, and the HIF-1α/BNIP3 signaling pathway was important in the
activation of hypoxia-induced autophagy in ACC. Furthermore,
inhibition of autophagy suppressed tumor invasion induced by
hypoxia. These findings reinforce the importance of autophagy in
tumor progression and indicate that manipulation of hypoxia-induced
autophagy may offer a novel target for ACC therapy.
Acknowledgments
The present study was supported by grants from the
Shandong Provincial Science and Technology Development Plan (grant
no. 2006GG2202034), the Science and Technology Foundation of the
Shandong Province (grant no. 2006GG20002046) and the Shandong
Provincial Natural Science Foundation (grant no. ZR2012HQ022).
References
|
1
|
Adelstein DJ, Koyfman SA, El-Naggar AK and
Hanna EY: Biology and management of salivary gland cancers. Semin
Radiat Oncol. 22:245–253. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dodd RL and Slevin NJ: Salivary gland
adenoid cystic carcinoma: A review of chemotherapy and molecular
therapies. Oral Oncol. 42:759–769. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bradley PJ: Adenoid cystic carcinoma of
the head and neck: A review. Curr Opin Otolaryngol Head Neck Surg.
12:127–132. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Janssen HL, Haustermans KM, Balm AJ and
Begg AC: Hypoxia in head and neck cancer: How much, how important?
Head Neck. 27:622–638. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rouschop KM and Wouters BG: Regulation of
autophagy through multiple independent hypoxic signaling pathways.
Curr Mol Med. 9:417–424. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Toustrup K, Sørensen BS, Alsner J and
Overgaard J: Hypoxia gene expression signatures as prognostic and
predictive markers in head and neck radiotherapy. Semin Radiat
Oncol. 22:119–127. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Adams JM, Difazio LT, Rolandelli RH, Luján
JJ, Haskó G, Csóka B, Selmeczy Z and Németh ZH: HIF-1: A key
mediator in hypoxia. Acta Physiol Hung. 96:19–28. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Semenza GL: HIF-1 and mechanisms of
hypoxia sensing. Curr Opin Cell Biol. 13:167–171. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brennan PA, Mackenzie N and Quintero M:
Hypoxia-inducible factor 1alpha in oral cancer. J Oral Pathol Med.
34:385–389. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Costa AF, Tasso MG, Mariano FV, Soares AB,
Chone CT, Crespo AN, Fresno MF, Llorente JL, Suárez C, de Araújo
VC, et al: Levels and patterns of expression of hypoxia-inducible
factor-1α, vascular endothelial growth factor, glucose
transporter-1 and CD105 in adenoid cystic carcinomas with
high-grade transformation. Histopathology. 60:816–825. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou C, Liu J, Tang Y, Zhu G, Zheng M,
Jiang J, Yang J and Liang X: Coexpression of hypoxia-inducible
factor-2α, TWIST2 and SIP1 may correlate with invasion and
metastasis of salivary adenoid cystic carcinoma. J Oral Pathol Med.
41:424–431. 2012. View Article : Google Scholar
|
|
12
|
Chen N and Karantza V: Autophagy as a
therapeutic target in cancer. Cancer Biol Ther. 11:157–168. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
White E and DiPaola RS: The double-edged
sword of autophagy modulation in cancer. Clin Cancer Res.
15:5308–5316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang L, Huang S, Li W, Zhang D, Zhang S,
Zhang W, Zheng P and Chen Z: Expression of autophagy and ER
stress-related proteins in primary salivary adenoid cystic
carcinoma. Pathol Res Pract. 208:635–641. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang L, Huang S, Zhang D, Zhang B, Li K,
Li W, Zhang S, Zhang W and Zheng P: Inhibition of autophagy
augments chemotherapy in human salivary adenoid cystic carcinoma. J
Oral Pathol Med. 43:265–272. 2014. View Article : Google Scholar
|
|
16
|
Ma B, Liang LZ, Liao GQ, Liang YJ, Liu HC,
Zheng GS and Su YX: Inhibition of autophagy enhances cisplatin
cytotoxicity in human adenoid cystic carcinoma cells of salivary
glands. J Oral Pathol Med. 42:774–780. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mazure NM and Pouysségur J:
Hypoxia-induced autophagy: Cell death or cell survival? Curr Opin
Cell Biol. 22:177–180. 2010. View Article : Google Scholar
|
|
19
|
Bellot G, Garcia-Medina R, Gounon P,
Chiche J, Roux D, Pouysségur J and Mazure NM: Hypoxia-induced
autophagy is mediated through hypoxia-inducible factor induction of
BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol.
29:2570–2581. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Azad MB and Gibson SB: Role of BNIP3 in
proliferation and hypoxia-induced autophagy: Implications for
personalized cancer therapies. Ann N Y Acad Sci. 1210:8–16. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang GL and Semenza GL: General
involvement of hypoxia-inducible factor 1 in transcriptional
response to hypoxia. Proc Natl Acad Sci USA. 90:4304–4308. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Song ZC, Zhou W, Shu R and Ni J: Hypoxia
induces apoptosis and autophagic cell death in human periodontal
ligament cells through HIF-1α pathway. Cell Prolif. 45:239–248.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Naves T, Jawhari S, Jauberteau MO,
Ratinaud MH and Verdier M: Autophagy takes place in mutated p53
neuroblastoma cells in response to hypoxia mimetic CoCl(2). Biochem
Pharmacol. 85:1153–1161. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rovetta F, Stacchiotti A, Faggi F,
Catalani S, Apostoli P, Fanzani A and Aleo MF: Cobalt triggers
necrotic cell death and atrophy in skeletal C2C12 myotubes. Toxicol
Appl Pharmacol. 271:196–205. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chinnadurai G, Vijayalingam S and Gibson
SB: BNIP3 subfamily BH3-only proteins: Mitochondrial stress sensors
in normal and pathological functions. Oncogene. 27(Suppl l):
S114–S127. 2008. View Article : Google Scholar
|
|
26
|
Vengellur A and LaPres JJ: The role of
hypoxia inducible factor 1α in cobalt chloride induced cell death
in mouse embryonic fibroblasts. Toxicol Sci. 82:638–646. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao X, He Y and Chen H: Autophagic tumor
stroma: Mechanisms and roles in tumor growth and progression. Int J
Cancer. 132:1–8. 2013. View Article : Google Scholar
|
|
28
|
Bohensky J, Shapiro IM, Leshinsky S,
Terkhorn SP, Adams CS and Srinivas V: HIF-1 regulation of
chondrocyte apoptosis: Induction of the autophagic pathway.
Autophagy. 3:207–214. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Maiuri MC, Le Toumelin G, Criollo A, Rain
JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K,
Tavernarakis N, et al: Functional and physical interaction between
Bcl-X (L) and a BH3-like domain in Beclin-1. EMBO J. 26:2527–2539.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Maiuri MC, Criollo A, Tasdemir E, Vicencio
JM, Tajeddine N, Hickman JA, Geneste O and Kroemer G: BH3-only
proteins and BH3 mimetics induce autophagy by competitively
disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X (L).
Autophagy. 3:374–376. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wouters BG, Weppler SA, Koritzinsky M,
Landuyt W, Nuyts S, Theys J, Chiu RK and Lambin P: Hypoxia as a
target for combined modality treatments. Eur J Cancer. 38:240–257.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Macintosh RL, Timpson P, Thorburn J,
Anderson KI, Thorburn A and Ryan KM: Inhibition of autophagy
impairs tumor cell invasion in an organotypic model. Cell Cycle.
11:2022–2029. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li J, Yang B, Zhou Q, Wu Y, Shang D, Guo
Y, Song Z, Zheng Q and Xiong J: Autophagy promotes hepatocellular
carcinoma cell invasion through activation of
epithelial-mesenchymal transition. Carcinogenesis. 34:1343–1351.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yamanaka-Tatematsu M, Nakashima A, Fujita
N, Shima T, Yoshimori T and Saito S: Autophagy Induced by HIF1α
over-expression supports trophoblast invasion by supplying cellular
energy. PloS One. 8:e766052013. View Article : Google Scholar
|
|
35
|
Gu BJ and Wiley JS: Rapid ATP-induced
release of matrix metal-loproteinase 9 is mediated by the P2X7
receptor. Blood. 107:4946–4953. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Indelicato M, Pucci B, Schito L, Reali V,
Aventaggiato M, Mazzarino MC, Stivala F, Fini M, Russo MA and
Tafani M: Role of hypoxia and autophagy in MDA-MB-231 invasiveness.
J Cell Physiol. 223:359–368. 2010.PubMed/NCBI
|