Introduction
Alzheimer's disease (AD) is a fatal incurable
neurodegenerative disease that is prevalent worldwide (1). The progressive disease is
characterized by the accumulation of plaques formed of short
β-amyloid (Aβ) peptides, neurofibrillary tangles inside neurons,
loss of neurons in the hippocampus and cerebral cortex, and
widespread brain atrophy. AD is the most common form of dementia in
the elderly, accounting for 50–70% of the late-onset dementia, with
17–20 million affected worldwide (2). Although several key proteins, such as
amyloid precursor protein (APP), β-secretase 1, Tau and
presenillin-1 (PSEN1), have been identified to be implicated in AD
pathogenesis, the etiology and pathogenesis of AD are not well
understood.
MicroRNAs are a group of small (~22 nucleotides)
endogenous noncoding RNAs, that function as suppressors of gene
expression. By binding to the 3′-UTR of their target genes, miRNAs
can induce mRNA degradation or translational repression (3,4).
miRNAs are abundant in the brain and crucially important in
neurodevelopment and synaptic plasticity (5,6).
Increasing evidence has revealed that alterations in miRNA
expression may contribute to the initiation and progression of AD
(7–11). However, the mechanism underlying
the role of microRNAs in AD has not been fully elucidated.
p27Kip1 is encoded by cyclin-dependent
kinase inhibitor 1B and functions to negatively control cell cycle
progression (12,13). Expression of p27Kip1 can
normally inhibit the phosphorylation of pRb and therefore arrest
cell proliferation at G1. A number of studies have shown that
disordered expression of cell-cycle markers, including
p27Kip1, contributes to the pathogenesis of AD (14,15).
However, p27Kip1 gene expression regulation is not
completely understood. The present study hypothesized that the
expression of p27Kip1 may be regulated by certain
miRNAs, and aimed to elucidate the interactions between miR-222 and
p27Kip1 in AD
Materials and methods
Animal model
The APPswe/PSΔE9 mice model was purchased from the
Jackson Laboratory (Ben Harbor, ME, USA). Briefly, the transgenic
mice were established by co-injection of the APPswe and PS1ΔE9
vectors (16). The expression of
the transgenes is under the control of the mouse prion protein
promoter, which can drive high protein expression in the neurons
and astrocytes of the central nervous system (17,18).
The APPswe transgene encodes a mouse-human hybrid transgene
containing the mouse sequence in the extracellular and
intracellular regions, and a human sequence within the Aβ domain
with Swedish mutations K594N/M595L. The PS1ΔE9 transgene encodes
the exon-9-deleted human PSEN1. Polymerase chain reaction (PCR)
amplification of tail DNA was performed to genotype the mice for
the presence of the transgene. The PCR reaction mixture of 20
µl contained 100 ng genomic DNA, 2 µl 10X buffer, 0.4
µl dNTPs (10 mM), 0.5 µl forward and reverse primers
(20 µM), and 0.2 U Taq DNA polymerase (Sangon
Biotech, Shanghai, China). The PCR was performed with
pre-denaturation at 95°C for 3 min, followed by 35 cycles of
denaturation at 95°C for 30 s, annealing at 54°C for 30 s (for APP)
or 52°C for 40 s (for PS1), elongation at 72°C for 30 s (for APP)
or 60 s (for PS1), and a final extension at 72°C for 10 min. Primer
sequences were as follows: Forward: 5′-GACTGACCACTCGACCAGGTTCTG-3′
and reverse: 5′-CTTGTAAGTTGGATTCTCATATCCG-3′ for APP (product
length, 344 bp); and forward: 5′-AATAGAGAACGG CAGGAGCA-3′ and
reverse: 5′-GCCATGAGGGCACTAATCAT-3′ for presenilin 1 (product
length, 608 bp). The transgenic mice and age-matched controls were
housed under controlled illumination (12 h light/dark cycle) in a
specific pathogen-free environment (room temperature, 20–22°C;
humidity, 55–60%). Food and water were available ad libitum.
Eight six month-old transgenic mice and eight age-matched controls
were used in this study. The mice were sacrificed by inhalation of
5% isoflurane (RWD Life Science, Shenzhen, China) prior to cervical
dislocation. Using small scissors, the head was severed and the
skin separated in order to fracture the skull. Using small
tweezers, small sections of skull were carefully removed in order
to expose the brain. The brain tissue samples were immediately
frozen in liquid nitrogen, and stored at −80°C until protein and
RNA extraction was performed. All animal protocols were conducted
in accordance with the Guide for the Care and Use of Laboratory
Animals of the National Institutes of Health (19). Formal approval of this study was
obtained from the Committee on the Ethics of Animal Experiments at
The Second Hospital of Shandong University (Jinan, China).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA from brains and cultured cells was
isolated using the mirVanaTM miRNA Isolation kit (AM1560, Ambion,
Austin, TX, USA) in accordance with the manufacturer's
instructions. The RNA was treated with DNase I (AM1906, Ambion) to
eliminate genomic DNA contamination. RNA (1 µg) was reverse
transcribed into miRNA cDNA and total cDNA using the miScript
Reverse Transcription kit (218061, Qiagen, Hilden, Germany),
followed by qPCR on a Lightcycler (Roche Diagnostics, Mannheim,
Germany) machine using the miScript SYBR Green PCR kit (218073,
Qiagen), according to the manufacturer's instructions. Primers for
mature miR-222 and U6snRNA were purchased from Qiagen (MS00007609
and MS00007497, Hilden, Germany). The primer sequence were as
follows: Forward: 5′-CAGGTCTCCAAGACGACATAGA-3′ and reverse:
5′-CGCCTTTTCGATTCATGTACTGC-3′ for p27Kip1 (20); and forward:
5′-CATGGGTGTGAACCATGAGA-3′ and reverse: 5′-TGTGGTCATGAGTCCTTCCA-3′
for GAPDH. All reactions were run in triplicate. Relative
expression levels for p27Kip1 mRNA and miR-222 were
determined using the 2−ΔΔCq method.
Cell culture
The SH-SY5Y and HEK-293T cell lines were obtained
from the Cell Bank of Chinese Academy of Sciences (Shanghai, China)
and routinely cultured in Dulbecco's modified Eagle's medium
(11965-092, Invitrogen Life Technologies, Carlsbad, CA, USA)
supplemented with 10% heat-inactivated fetal bovine serum
(Invitrogen Life Technologies), 100 U/ml penicillin and 100
µg/ml streptomycin (Sigma-Aldrich, St. Louis, MO, USA) in a
humidified air atmosphere of 5% CO2 at 37°C.
miR-222 overexpression in cultured
cells
Overexpression of miR-222 was performed using an
miR-222 expression vector, which was constructed using BLOCK-iTTM
Pol II miR RNAi Expression Vector kit with EmGFP (K4936-00,
Invitrogen Life Technologies) according to the manufacturer's
instructions. The top oligo sequence for miR-222 is
5′-TGCTGAGCTACATCTGGCTACTGGGTGTTTTGGCCACTGACTGACACCCAGTACAGATGTAGCT-3′,
and the bottom oligo sequence for miR-222 is
5′-CCTGAGCTACATCTGTACTGGGTGTCAGTCAGTGGCCAAAACACCCAGTAGCCAGATGTAGCTC-3′.
The negative control vector was provided by Invitrogen Life
Technologies, which contains an insert that can form a hairpin
structure just as a regular pre-miRNA, but is predicted not to
target any known vertebrate gene. The expression vector or control
vector was transfected into the SH-SY5Y cell line with
Lipofectamine 2000 reagent (11668-019, Invitrogen Life
Technologies). To generate the stable cell line that can
constitutively express miR-222, blasticidin (15205, Sigma-Aldrich)
was added 24 h after transfection at concentration of 3
µg/ml for 10 days. Resistant cells were analyzed by
fluorescent microscopy (TE2000-U; Nikon Corporation, Tokyo,
Japan).
miR-222 knockdown in cultured cells
The inhibition of miR-222 in SH-SY5Y cells was
produced with the locked nucleic acid (LNA) oligonucleotides for
specific in vitro knockdown. The LNA oligonucleotides
(exhibiting high affinity to complementary nucleic acids and able
to efficiently silence their target miRNAs in vitro and
in vivo) were purchased from Exiqon (Vedbaek, Denmark) and
transfected with Lipofectamine 2000 reagent (11668-019, Invitrogen
Life Technologies, Carlsbad, CA, USA) at a final concentration of
80 nM. The cells were collected 48 h after transfection, and the
levels of miR-222 and p27Kip1 were analyzed.
Dual-luciferase reporter assay
The analysis by the miRNA target prediction
algorithm TargetScan (http://www.targetscan.org/) revealed that
p27Kip1 is a candidate target gene of miR-222. The
3′-UTR of human p27Kip1 was amplified by PCR using the
following primers 5′-TAAGAATATGTTTCCTTGTTTATCAGAT-3′ and
5′-AATAGCTATGGAAGTTTTCTTTATTGAT-3′. The PCR products were
directionally cloned downstream of the Renilla luciferase
stop codon in the psiCHECKTM-2 vector (C-8021, Promega Corporation,
Madison, WI, USA). The 3′-UTR of the mutant vector of
p27Kip1 was also constructed by the overlap extension
PCR method using the following primers:
5′-CTCTAAAAGCGTTGGAGCATTATGCAATTAGG-3′ and
5′-CCTAATTGCATAATGCTCCAACGCTTTTAGAG-3′. The wild-type or mutant
p27Kip1 3′-UTR psiCHECK-2 plasmid was transiently
co-transfected with miR-222 expression vector or negative control
vector into HEK-293T cells. Cell lysates were harvested 48 h after
transfection and then firefly and Renilla luciferase was
measured with the Dual Luciferase Reporter Assay system (E1910,
Promega Corporation) following the manufacturer's instructions.
Renilla luciferase activities were normalized to firefly
luciferase activities to also control transfection efficiency.
Western blot analysis
The cells and tissues were homogenized in chilled
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology, Shanghai, China) and stood for 30 min on ice. After
centrifugation at 17,530 × g for 30 min at 4°C, the supernatants
were collected as protein samples. The protein concentration was
detected by the BCATM Protein Assay kit (Pierce, Biotechnology
Inc., Rockford, IL, USA). Proteins (40 µg) were separated on
12% SDS-PAGE gels (Thermo Fisher Scientific, Inc., Waltham, MA,
USA), and transferred to nitrocellulose membranes (Millipore,
Billerica, MA, USA). The membranes were blocked with 5% non-fat
milk for 2 h, and incubated overnight at 4°C with mouse monoclonal
anti-human/mouse primary antibody against p27Kip1
(1:250; cat. no. ab82264, Abcam, Cambridge, MA, USA) and rabbit
polyclonal anti-human/mouse primary antibody against GAPDH (1:500;
cat. no. D261392; Sangon Biotech). The membranes were then washed
in Tris-buffered saline (TBS) for three times, and incubated for 1
h at room temperature with horseradish peroxidase-conjugated goat
anti-mouse/rabbit secondary antibody (1:1,000; cat. no.
A0216/A0208; Beyotime Institute of Biotechnology). Subsequent to
another three washes in TBS, signals from the bound secondary
antibody were generated using the enhanced chemiluminescence
solution (Amersham, Piscataway, NJ, USA) and were detected by
exposure of the membranes to X-ray films (Kodak, Rochester, NY,
USA). The relative signal intensity was quantified by densitometry
with UVIPhoto and UVISoft UVIBand Application V97.04 (Uvitech,
Cambridge, UK).
Statistical analysis
Statistical analyses were performed with the
statistical software package, SPSS, version 17.0 (SPSS, Inc.,
Chicago, IL, USA). The results are expressed as the mean ± standard
deviation of three independent experiments. The differences between
groups were analyzed by Student's t-test. P-values are two-sided,
and P≤0.05 was considered to indicate a statistically significant
difference.
Results
miR-222 is downregulated in APPswe/PSΔE9
mice versus control mice
Eight 6-month-old transgenic mice and eight
age-matched controls were used in this study. RT-qPCR was performed
to evaluate the miR-222 expression level in 8 APPswe/PSΔE9 mice and
8 controls. All reactions were run in triplicate. As shown in
Fig. 1A, there was a significantly
decreased level of miR-222 in the APPswe/PSΔE9 mice compared with
the control group.
Reduced miR-222 is correlated with high
level of p27Kip1 protein in APPswe/PSΔE9 mice and
control mice
To investigate the potential interaction of miR-222
and p27Kip1 in vivo, western blot analysis was
performed on the total protein extracts from the cerebral cortex of
6-month-old APPswe/PSΔE9 mice and controls. It was demonstrated
that the level of p27Kip1 protein was increased in the
APPswe/PSΔE9 mice compared with age-matched controls (Fig. 1B and C), which is inversely
associated with the expression of miR-222. In addition, the mRNA
level of p27Kip1 was also detected by RT-qPCR in the
same mice and no significant difference was observed (Fig. 1D). It is well-known that miRNAs
execute post-transcriptional regulation by inducing mRNA
degradation or translational repression. The present data indicated
that low miR-222 expression may positively regulate
p27Kip1 expression by promoting the translation of
p27Kip1 mRNA.
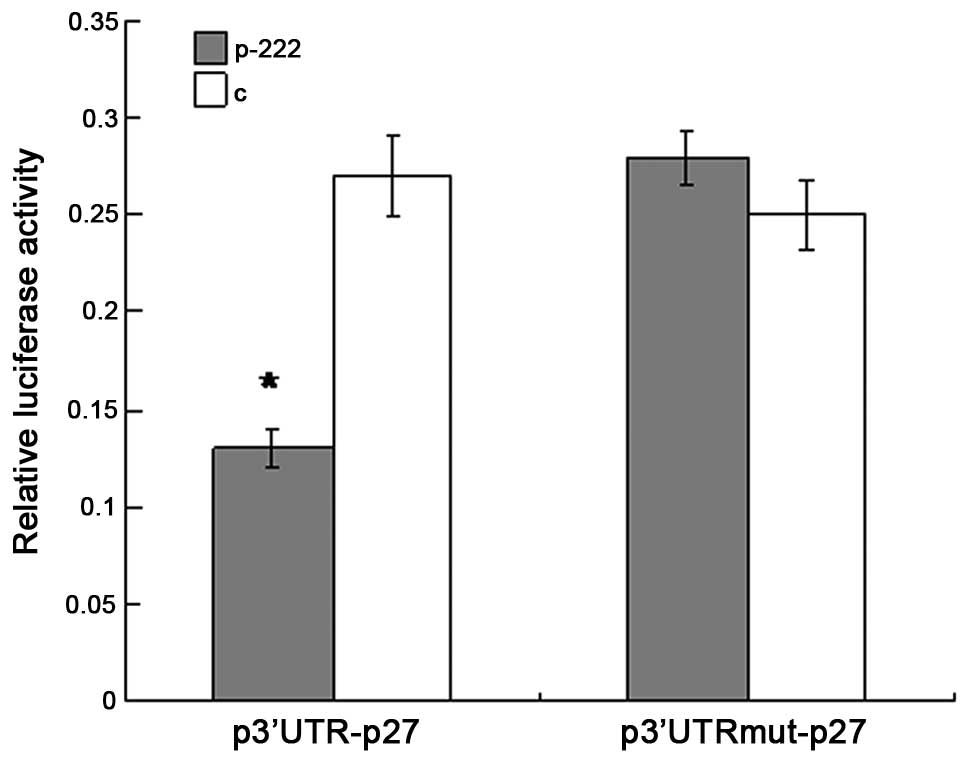
3′UTR of p27Kip1 mRNA is
targeted by miR-222 in HEK-293T cells
To investigate whether p27Kip1 is a
target gene of miR-222 the alignment of miR-222 with the 3′-UTR of
p27Kip1 was analyzed by the miRNA target prediction
algorithm TargetScan (http://www.targetscan.org/). It was shown that there
was one putative binding site in the 3′-UTR of p27Kip1,
which has conserved binding sites for miR-222. To experimentally
validate the potential interaction, a luciferase reporter con
taining the 3′-UTR of human p27Kip1 was cotransfected
into HEK-293T cells with the miR-222-expression vector or negative
control vector. The luciferase activity was measured 48 h post
transfection. As shown in Fig. 2,
the overexpression of miR-222 produced a highly significant
reduction (54%, P<0.05) in luciferease activity of the reporter
plasmid compared with the controls. In addition, when the predicted
binding site of miR-222 in the 3′-UTR was mutated by overlapping
PCR, the repression of luciferease activity caused by miR-222
overexpression was abrogated. Collectively, the data suggest that
p27Kip1 is a predicted target of miR-222, and the
inhibitory effect of miR-222 is due to direct interaction with the
putative binding site of the 3′-UTR of p27Kip1.
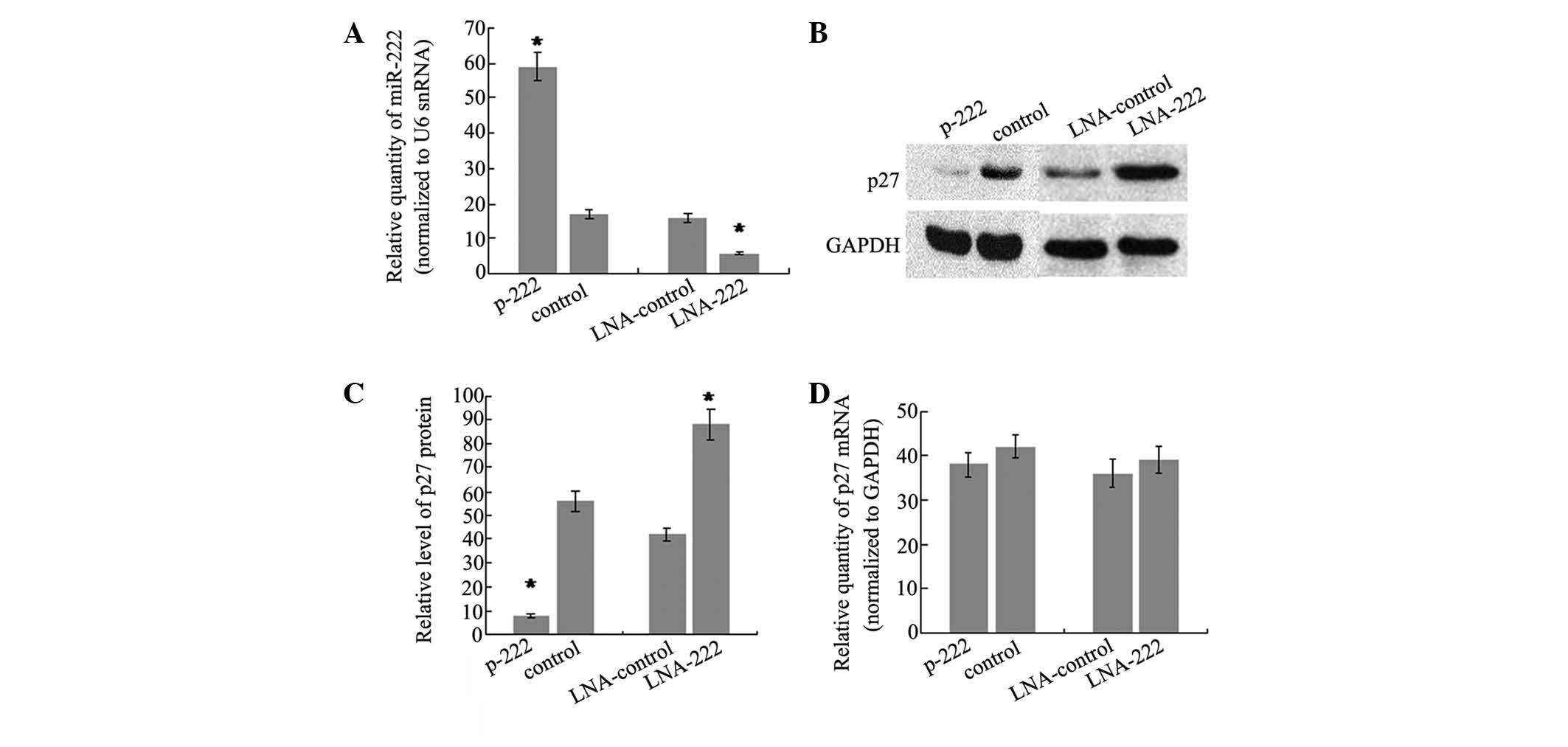
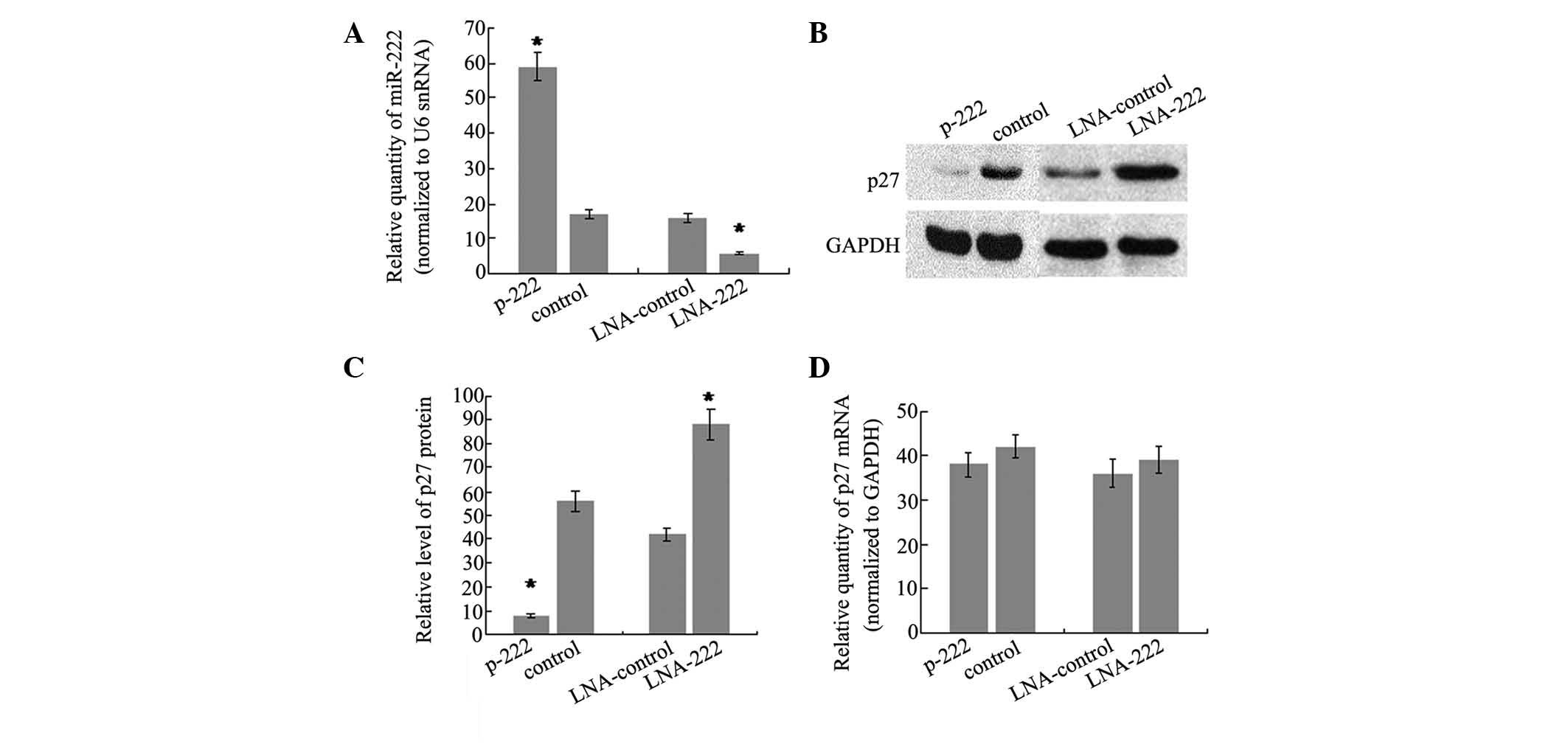
miR-222 regulates p27Kip1
expression in SH-SY5Y cells
To further determine whether miR-222 can directly
regulate p27Kip1, the changes in the p27Kip1
level in SH-SY5Y cells following miR-222 overexpression were
analyzed. To do this, the stable transfectant cell line of miR-222,
which showed a 3.5-fold increase in miR-222 levels compared with
controls (Fig. 3A), was generated.
In addition, western blot analysis was performed on the same cells
to evaluate the protein level of p27Kip1. The results
showed that there was a significant reduction in the
p27Kip1 protein level (86%, P<0.05) in the stable
transfection cell line of miR-222, as compared with cells
transfected with the control vector (Fig. 3B and C). Furthermore, miR-222
expression was knocked down by trans-fecting SH-SY5Y cells with LNA
antisense oligonucleotides targeting miR-222 and the effects on
p27Kip1 production were analyzed. There was a 2.7-fold
decrease of miR-222 level in SH-SY5Y cells transfected with
anti-222 LNA compared with cells transfected with LNA against a
microRNA not expressed in these cells (Fig. 3A). As expected, the reduction of
miR-222 was accompanied by an increase in p27Kip1
protein of 2.1-fold (Fig. 3B and
3C). It is well-understood that
repression of gene expression by miRNAs may be due to translational
repression or mRNA degradation. To test the relative contribution
of the two mechanisms, the p27Kip1 mRNA level was
detected and no significant difference was observed following
overexpression or knockdown of miR-222. The data demonstrated that
miR-222 may regulate p27Kip1 expression by interfering
with translation of p27Kip1 mRNA, rather than inducing
mRNA degradation.
Discussion
miRNAs are small non-coding RNA molecules, ~ 22
nucleotides in length, that regulate gene expression by repressing
translation or degrading target mRNAs. miRNAs are abundantly
expressed in the brain, where they regulate diverse neuronal and
glial functions (4). Numerous
types of miRNAs have been recognized to be correlated with
neurodegenerative diseases (21–23).
miR-222 is a member of the to the miR-221/222 family and has been
identified to be overexpressed in several types of cancer (24–27).
Growing evidence has indicated the important roles of miR-222 in
tumor proliferation, drug resistance, apoptosis and metastasis
(28–30). However, for miR-222, the possible
roles and associated target genes in AD is unclear. In the current
study, it was observed that miR-222 is downregulated in
APPswe/PSΔE9 mice compared with age-matched controls. In addition,
the overexpression of miR-222 or its knockdown produced the
predictable opposite effects on p27Kip1 expression.
Through the luciferase assay, it was demonstrated that miR-222 can
directly target p27Kip1 by interaction with the putative
binding site of the 3′-UTR of p27Kip1.
Previous studies have reported that the
dysregulation of cell-cycle markers is important in AD pathogenesis
(31–39). p27Kip1, also termed
cyclin-dependent kinase inhibitor 1B, can prevent cell-cycle
progression from the G1 to S phase by binding to CDK2 and cyclin E
complexes. Extensive evidence has suggested that the overexpression
of p27Kip1 is correlated with the pathogenesis of AD.
However, p27Kip1 gene expression regulation is not
completely understood. The present study analyzed the expression of
p27Kip1 in the cerebral cortex of APPswe/PSΔE9 mice and
demonstrated that the protein level of p27Kip1 is
upregulated in these mice compared with age-matched controls.
However, there was no significant difference in mRNA level of
p27Kip1 between the model mice and controls. This
indicates that p27Kip1 mRNA may be regulated through a
post-transcriptional mechanism. Sequence analysis by Targetscan
suggested that the 3′-UTR of p27Kip1 mRNA contained a
predicted binding site for miR-222. An inverse correlation between
the expression of miR-222 and p27Kip1 was identified in
the model mice and controls. Additionally, there was an inverse
correlation between the p27Kip1 and miR-222 expression
levels in SH-SY5Y cells. Collectively, the results indicated that
p27Kip1 is a direct target of miR-222 and the
downregulation of miR-222 may contribute to the dysregulation of
the cell cycle in AD due to dysregulation of the expression of
p27Kip1.
In conclusion, it was confirmed that miR-222 was
down-regulated in AD, in association with p27Kip1
upregulation. miR-222 can directly target p27Kip1 by
binding to the 3′-UTR of p27Kip1 inducing the
translational repression of p27Kip1. Therefore,
downregulation of miR-222 may be correlated with the dysregulation
of the cell cycle in AD by affecting the expression of
p27Kip1.
Acknowledgments
The authors would like to thank Dr Wei Tong and Dr
Xueli Liu for technical assistance, and Dr Jiamei Li, Dr Lingling
Zhang, Miss Haixia Shi and Miss Xiaoying Li for their help with
research. The present study was supported by grants from the
Shandong Provincial Natural Science Foundation of China (grant no.
ZR2015PH038).
References
|
1
|
Cornutiu G: The Epidemiological Scale of
Alzheimer's Disease. J Clin Med Res. 7:657–666. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Blennow K, de Leon MJ and Zetterberg H:
Alzheimer's disease. Lancet. 368:387–403. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim VN: Small RNAs: Classification,
biogenesis and function. Mol Cells. 19:1–15. 2005.PubMed/NCBI
|
|
4
|
Petersen CP, Bordeleau ME, Pelletier J and
Sharp PA: Short RNAs repress translation after initiation in
mammalian cells. Mol Cell. 21:533–542. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hébert SS and De Strooper B: Molecular
biology. miRNAs in neurodegeneration. Science. 317:1179–1180. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kosik KS: The neuronal microRNA system.
Nat Rev Neurosci. 7:911–920. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lukiw WJ: Micro-RNA speciation in fetal,
adult and Alzheimer's disease hippocampus. Neuroreport. 18:297–300.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang WX, Rajeev BW, Stromberg AJ, Ren N,
Tang G, Huang Q, Rigoutsos I and Nelson PT: The expression of
microRNA miR-107 decreases early in Alzheimer's disease and may
accelerate disease progression through regulation of beta-site
amyloid precursor protein-cleaving enzyme 1. J Neurosci.
28:1213–1223. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Krichevsky AM, King KS, Donahue CP,
Khrapko K and Kosik KS: A microRNA array reveals extensive
regulation of microRNAs during brain development. RNA. 9:1274–1281.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hébert SS and De Strooper B: Alterations
of the microRNA network cause neurodegenerative disease. Trends
Neurosci. 32:199–206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nelson PT, Wang WX and Rajeev BW:
MicroRNAs (miRNAs) in neurodegenerative diseases. Brain Pathol.
18:130–138. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Polyak K, Lee MH, Erdjument-Bromage H,
Koff A, Roberts JM, Tempst P and Massagué J: Cloning of
p27Kip1, a cyclin-dependent kinase inhibitor and a
potential mediator of extracellular antimitogenic signals. Cell.
78:59–66. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Toyoshima H and Hunter T: p27, a novel
inhibitor of G1 cyclin-Cdk protein kinase activity, is related to
p21. Cell. 78:67–74. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ogawa O, Lee HG, Zhu X, Raina A, Harris
PL, Castellani RJ, Perry G and Smith MA: Increased p27, an
essential component of cell cycle control, in Alzheimer's disease.
Aging Cell. 2:105–110. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Delobel P, Lavenir I, Ghetti B, Holzer M
and Goedert M: Cell-cycle markers in a transgenic mouse model of
human tauopathy: Increased levels of cyclin-dependent kinase
inhibitors p21Cip1 and p27Kip1. Am J Pathol.
168:878–887. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jankowsky JL, Fadale DJ, Anderson J, Xu
GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner
SL, et al: Mutant presenilins specifically elevate the levels of
the 42 residue beta-amyloid peptide in vivo: Evidence for
augmentation of a 42-specific gamma secretase. Hum Mol Genet.
13:159–170. 2004. View Article : Google Scholar
|
|
17
|
Borchelt DR, Davis J, Fischer M, Lee MK,
Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia
SS and Price DL: A vector for expressing foreign genes in the
brains and hearts of transgenic mice. Genet Anal. 13:159–163. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lesuisse C, Xu G, Anderson J, Wong M,
Jankowsky J, Holtz G, Gonzalez V, Wong PC, Price DL, Tang F, et al:
Hyper-expression of human apolipoprotein E4 in astroglia and
neurons does not enhance amyloid deposition in transgenic mice. Hum
Mol Genet. 10:2525–2537. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mason TJ and Matthews M: Aquatic
environment, housing, and management in the eighth edition of the
Guide for the Care and Use of Laboratory Animals: Additional
considerations and recommendations. J Am Assoc Lab Anim Sci.
51:329–332. 2012.PubMed/NCBI
|
|
20
|
Yang YF, Wang F, Xiao JJ, Song Y, Zhao YY,
Cao Y, Bei YH and Yang CQ: MiR-222 overexpression promotes
proliferation of human hepatocellular carcinoma HepG2 cells by
downregulating p27. Int J Clin Exp Med. 7:893–902. 2014.PubMed/NCBI
|
|
21
|
Kim J, Inoue K, Ishii J, Vanti WB, Voronov
SV, Murchison E, Hannon G and Abeliovich A: A MicroRNA feedback
circuit in midbrain dopamine neurons. Science. 317:1220–1224. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schaefer A, O'Carroll D, Tan CL, Hillman
D, Sugimori M, Llinas R and Greengard P: Cerebellar
neurodegeneration in the absence of microRNA. J Exp Med.
204:1553–1558. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bilen J, Liu N, Burnett BG, Pittman RN and
Bonini NM: MicroRNA pathways modulate polyglutamine-induced
neuro-degeneration. Mol Cell. 24:157–163. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Galardi S, Mercatelli N, Giorda E,
Massalini S, Frajese GV, Ciafrè SA and Farace MG: miR-221 and
miR-222 expression affects the proliferation potential of human
prostate carcinoma cell lines by targeting p27Kip1. J
Biol Chem. 282:23716–23724. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Greither T, Grochola LF, Udelnow A,
Lautenschläger C, Würl P and Taubert H: Elevated expression of
microRNAs 155, 203, 210 and 222 in pancreatic tumors is associated
with poorer survival. Int J Cancer. 126:73–80. 2010. View Article : Google Scholar
|
|
26
|
Visone R, Russo L, Pallante P, De Martino
I, Ferraro A, Leone V, Borbone E, Petrocca F, Alder H, Croce CM and
Fusco A: MicroRNAs (miR)-221 and miR-222, both overexpressed in
human thyroid papillary carcinomas, regulate p27Kip1
protein levels and cell cycle. Endocr Relat Cancer. 14:791–798.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Quintavalle C, Garofalo M, Zanca C, Romano
G, Iaboni M, del Basso De Caro M, Martinez-Montero JC, Incoronato
M, Nuovo G, Croce CM and Condorelli G: miR-221/222 over-expression
in human glioblastoma increases invasiveness by targeting the
protein phosphate PTPµ. Oncogene. 31:858–868. 2012. View Article : Google Scholar
|
|
28
|
Stinson S, Lackner MR, Adai AT, Yu N, Kim
HJ, O'Briene C, Spoerke J, Jhunjhunwala S, Boyd Z, Januario T, et
al: miR-221/222 targeting of trichorhinophalangeal 1 (TRPS1)
promotes epithelial-to-mesenchymal transition in breast cancer. Sci
Signal. 4:pt52011.PubMed/NCBI
|
|
29
|
Stinson S, Lackner MR, Adai AT, Yu N, Kim
HJ, O'Brien C, Spoerke J, Jhunjhunwala S, Boyd Z, Januario T, et
al: TRPS1 targeting by miR-221/222 promotes the
epithelial-to-mesenchymal transition in breast cancer. Sci Signal.
4:ra412011.PubMed/NCBI
|
|
30
|
Yang CJ, Shen WG, Liu CJ, Chen YW, Lu HH,
Tsai MM and Lin SC: miR-221 and miR-222 expression increased the
growth and tumorigenesis of oral carcinoma cells. J Oral Pathol
Med. 40:560–566. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Arendt T, Rödel L, Gärtner U and Holzer M:
Expression of the cyclin-dependent kinase inhibitor p16 in
Alzheimer's disease. Neuroreport. 7:3047–3049. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
McShea A, Harris PL, Webster KR, Wahl AF
and Smith MA: Abnormal expression of the cell cycle regulators P16
and CDK4 in Alzheimer's disease. Am J Pathol. 150:1933–1939.
1997.PubMed/NCBI
|
|
33
|
Busser J, Geldmacher DS and Herrup K:
Ectopic cell cycle proteins predict the sites of neuronal cell
death in Alzheimer's disease brain. J Neurosci. 18:2801–2807.
1998.PubMed/NCBI
|
|
34
|
Arendt T, Holzer M and Gärtner U: Neuronal
expression of cycline dependent kinase inhibitors of the INK4
family in Alzheimer's disease. J Neural Transm. 105:949–960. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tsujioka Y, Takahashi M, Tsuboi Y,
Yamamoto T and Yamada T: Localization and expression of cdc2 and
cdk4 in Alzheimer brain tissue. Dement Geriat Cogn Disord.
10:192–198. 1999. View Article : Google Scholar
|
|
36
|
Jordan-Sciutto KL, Malaiyandi LM and
Bowser R: Altered distribution of cell cycle transcriptional
regulators during Alzheimer disease. J Neuropathol Exp Neurol.
61:358–367. 2002.PubMed/NCBI
|
|
37
|
Hoozemans JJ, Bruckner MK, Rozemuller AJ,
Veerhuis R, Eikelenboom P and Arendt T: Cyclin D1 and cyclin E are
co-localized with cyclooxygenase 2 (COX-2) in pyramidal neurons in
Alzheimer disease temporal cortex. J Neuropathol Exp Neurol.
61:678–688. 2002.PubMed/NCBI
|
|
38
|
Nagy Z, Esiri MM, Cato AM and Smith AD:
Cell cycle markers in the hippocampus in Alzheimer's disease. Acta
Neuropathol. 94:6–15. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ding XL, Husseman J, Tomashevski A,
Nochlin D, Jin LW and Vincent I: The cell cycle Cdc25A tyrosine
phosphatase is activated in degenerating postmitotic neurons in
Alzheimer's disease. Am J Pathol. 157:1983–1990. 2000. View Article : Google Scholar : PubMed/NCBI
|