Introduction
Hepatocellular carcinoma (HCC) is one of the most
common types of malignancy in China (1, 2).
HCC presents a serious threat to human health due to its rising
incidence, high metastatic recurrence and mortality rate (3, 4).
Although numerous treatment options exist, including surgical
resection and chemotherapy, the prognosis of HCC remains poor
(5). Identifying novel molecular
targets, developing novel drugs and researching mechanism-based
agents are required for the improvement of HCC treatment.
Cannabis sativa has been used medically for
several centuries. Cannabinoids are the major effective compound
present in Cannabis sativa. Numerous previous studies have
demonstrated that cannabinoids exert cell growth inhibition and
antitumor effects (6–11). Furthermore, the cannabinoid
receptors, which consist of seven transmembrane spanning domains,
have been cloned. Two cannabinoid receptors have been identified to
date: Cannabinoid receptor 1 (CB1) and 2 (CB2). A previous study
demonstrated that the cannabinoid, WIN55, 212-2 (WIN), inhibited
the proliferation of LNCap prostate cancer cells via cell cycle
arrest at the G0/G1 phase, and elucidated the underlying mechanism
(11). Furthermore, WIN has been
demonstrated to inhibit the cell cycle of the BEL7402 HCC cell
line; however, its underlying mechanism remains to be elucidated
(12). In addition, cannabinoids
have been reported to inhibit the metastasis of non-small cell lung
cancer (13). However, little is
currently known regarding the role of synthetic cannabinoids in
BEL7402 cell cycle and metastasis.
The present study demonstrated that treatment of
BEL7402 HCC carcinoma cells with the cannabinoid receptor agonist,
WIN, led to cell cycle arrest at the G0/G1 phase. Cell cycle arrest
was associated with inactivation of extracellular signal-regulated
kinases (ERK)1/2, increased expression of p27, and decreased
expression of cyclin D1 and cyclin-dependent kinase (Cdk)4.
Inhibiting CB2 with the CB2 antagonist, AM630, led to the
inactivation of ER K1/2. Inhibition of E R K1/2 signaling by its
inhibitor PD98059 also resulted in similar effects. The present
study also aimed to determine the role of WIN on BEL7402 cell
migration, and to explore the potential underlying mechanisms.
Materials and methods
Materials
R-(+)-[2,3-Dihydro-5-methyl-3[(4-morpholinyl)
methyl]pyrrolo[1,2,3-de]-1,4-benzoxazinyl]-(1-naphthalenyl)
methanone mesylate salt (WIN) and dimethyl sulfoxide (DMSO) were
purchased from Sigma-Aldrich (St. Louis, MO, USA). The CB2
antagonist, AM630, was purchased from Tocris Bioscience (Bristol,
UK). The CB2 selective agonist, JWH-015, was purchased from Enzo
Life Sciences, Inc. (Farmingdale, NY, USA). The mitogen-activated
protein kinase (MAPK) antagonist, PD98059, was purchased from
Beyotime Institute of Biotechnology (Haimen, China). Rat polyclonal
anti-CB2 antibodies were purchased from Abcam (Cambridge, MA, USA;
cat no. ab3561; 1:200 dilution). Rabbit polyclonal anti-matrix
metalloproteinase (MMP)9 antibodies were purchased from Rockland
Immunochemicals Inc. (Philadelphia, PA, USA; cat no. 600-401-CU9;
1:1,000 dilution). Rabbit polyclonal anti-cyclin D1 (cat no. SC753;
1:300 dilution) and mouse monoclonal CDK4 (cat no. SC23896; 1:1,000
dilution) antibodies were purchased from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA). Rabbit monoclonal phosphorylated (p)-p42/44
MAPK (ERK1/2) (Thr202/Tyr204) (cat no. 4094; 1:1,000 dilution) and
rabbit monoclonal p-retinoblastoma (Rb) (cat no. 8516; 1:1,000
dilution) antibodies were purchased from Cell Signaling Technology,
Inc. (Danvers, MA, USA). Rabbit polyclonal p27 (cat no. 25614-1-AP;
1:200 dilution), rabbit polyclonal E2F1 (cat no. 12334-1-AP; 1:300
dilution) and rabbit polyclonal β-actin (cat no. 20536-1-AP;
1:1,000 dilution) antibodies were purchased from Proteintech Group,
Inc. (Chicago, IL, USA).
Cell culture
BEL7402 cells (Institute of Biochemistry and Cell
Biology, Shanghai Institutes for Biological Sciences, Chinese
Academy of Sciences, Shanghai, China) were cultured in RPMI-1640
medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
supplemented with 10% (v/v) heat-inactivated fetal calf serum
(Zhejiang Tianhang Biotechnology Co., Ltd., Hangzhou, China), 2 mM
L-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin
(all from Beyotime Institute of Biotechnology), and incubated in a
humidified atmosphere containing 5% CO2.
Cell viability and anti-proliferation
assay
BEL7402 cells were seeded into 96-well plates at
density of 5×103 cells/well in 100 µl cell
medium. The cells were allowed to adhere for 24 h, and were
subsequently treated with PD98059 at 0, 5, 10, 20, 30 or 40
µm, or WIN at
0, 5, 10 or 20 µM for 24 h. Subsequently, 20 µl Cell
Counting kit-8 solution (Nanjing KeyGen Biotech Co., Ltd., Nanjing,
China) was added to each well and the culture was incubated for 1 h
at 37°C. All experiments were performed at least three times. The
optical density values were read at 450 nm using a microplate
reader (no. 680; Bio Rad Laboratories, Inc., Hercules, CA,
USA).
Cell treatment
WIN55, 212-2, dissolved in DMSO, was used to treat
the cells. For experiments, the cells were seeded at 60–70%
confluence, allowed to adhere overnight and subsequently treated
with the compounds. The final concentration of DMSO used was 0.1%
(v/v) for each treatment. For dose-dependent studies, BEL7402 cells
were treated with WIN at 0, 5 or 10 µm final concentration for 24 h in
serum-free medium. For the subsequent experiments, control cells
were treated with vehicle alone, and the WIN groups were treated
with 10 µm WIN
for 24 h. To explore the role of the CB2 receptor in WIN-induced
ERK1/2 inactivation, the cells were pretreated with 10 µM AM630 for
0.5 h followed by incubation with both 10 µM AM630 and 10 µM
WIN for 24 h. To study CB2 selective agonist JWH-015-induced ERK1/2
inactivation, the cells were treated with 4 µM JWH-015 for
24 h, and the WIN group was treated with 10 µM for 24 h. To
assess the role of ERK1/2 in cannabinoid receptor-induced cell
growth inhibition, the cells were treated with 30 µM ERK1/2
inhibitor, PD98059, for 24 h.
Flow cytometric cell cycle analysis
The cells were grown to a density of
1×106 cells in 100 mm culture dishes and were treated
for 24 h as described above. The cells were harvested by
trypsinization, re-suspended in phosphate-buffered saline (PBS) and
fixed in 70% (v/v) ethyl alcohol overnight at 4°C. Cell cycle
analysis was performed using a Cell Cycle kit (BestBio, Shanghai,
China), and cell cycle distribution was evaluated using the
FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ,
USA). The results were analyzed using ModFit LT software (version
3.2; Verity Software House, Topsham, ME, USA).
Cell migration assay
Cell migration was detected using Transwell
migration and wound healing assays. For the Transwell assay,
BEL7402 cells were treated with WIN at a concentration of 0, 5 or
10 µM for 24 h. The cells were subsequently trypsinized and
suspended in medium containing 2% fetal bovine serum (FBS). Cell
suspensions (200 µl), containing 2×105 cells,
were seeded into the upper chamber of a 24-well Transwell (pore
size, 8 µm; Corning Incorporated, Corning, NY, USA). The Transwells
were then inserted into a 24-well plate, containing 600 µl
RPMI-1640 medium, supplemented with 10% FBS and incubated at 37°C
in a humidified atmosphere for 24 h, in order to allow the BEL7402
cells to migrate. Cells on the upper side of the filter (not
migrated) were removed with cotton swabs. Migrated cells on the
lower side of the filter were fixed and stained with DAPI (Beyotime
Institute of Biotechnology). The number of BEL7402 cells that had
migrated to the lower surface of the membrane was counted in five
random and non-repeated high-power felds under a fluorescence
microscope (Nikon Eclipse Ti-S; Nikon, Tokyo, Japan). The average
number of migrated cells for each group was subsequently
calculated. Each assay was performed in triplicate wells. Cell
migration was also assessed using a scratch wound healing assay. To
visualize the migration of BEL7402 cells into artificial wounds,
the BEL7402 cells were seeded into 6-well plates and allowed to
grow for 24 h in RPMI-1640 medium, supplemented with 10% FBS. A 200
µl plastic pipette tip was used to gently scratch the cell
monolayer, in order to create a cell-free area. Subsequently, the
cells were washed extensively with PBS to remove cellular debris.
The cells were then incubated with 0, 5 or 10 µM WIN in
serum-free RPMI-1640 for 24 h. Wound closure was monitored after
staining with DAPI. Images of marked regions along the wounded area
were obtained using an inverted microscope (Nikon Eclipse Ti-S)
attached to a camera.
Western blot analysis
Once the cells were treated with the indicated
compounds, the total protein was extracted from the cells by
washing in PBS and incubating for 20 min in ice-cold lysis buffer,
supplemented with a protease and phosphatase inhibitor cocktail
(Roche Diagnostics Ltd., Shanghai, China). The cells were sonicated
three times for 10 sec and the protein concentrations were
subsequently determined using a bicinchoninic acid assay (Beyotime
Institute of Biotechnology). Equal quantities of protein samples
(80 µg/lane) were separated by 12% SDS-PAGE (SDS-PAGE kit;
Beijing Dingguochangsheng Biotechnology Co. Ltd., Beijing, China)
and then electrotransferred onto polyvinylidene fluoride membranes
(EMD Millipore, Bedford, MA, USA). Non-specific sites on the blots
were blocked by a 1 h incubation at room temperature with blocking
buffer (5% nonfat dry milk, 1% Tween 20 in 20 mmol/l Tris-buffered
saline; pH 7.6). The membranes were then incubated with the
specific primary antibodies overnight at 4°C, followed by a 1
hincubation at room temperature with horseradish
peroxidase-conjugated secondary antibodies (Proteintech Group,
Inc.). The blots were developed using enhanced chemiluminescence
reagents (Beyotime Institute of Biotechnology), according to the
manufacturer's protocol.
Statistical analysis
The results are expressed as the mean ± standard
deviation. The data were analyzed by Student's t-test in order to
determine statistical significance. Statistical analyses were
performed using SPSS 13.0 (SPSS, Inc., Chicago, IL, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
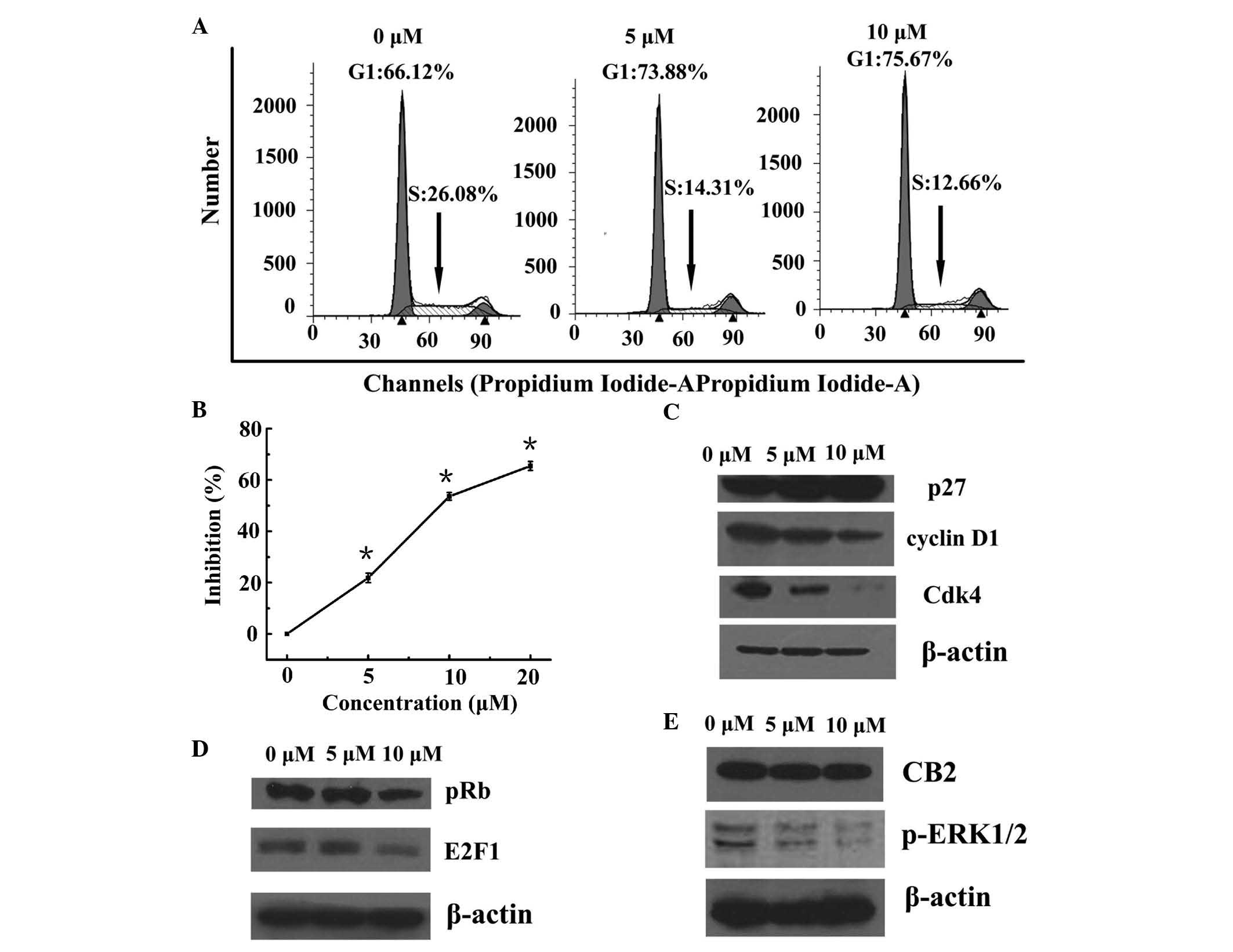
WIN results in cell cycle arrest at G0/G1
phase and inhibition of proliferation
The present study aimed to test the hypothesis that
WIN inhibited the proliferation of BEL7402 cells via cell cycle
arrest. A DNA cell cycle analysis was performed, in order to assess
the effects of WIN treatment on cell cycle distribution. As shown
in Fig. 1A, treatment with WIN
resulted in a dose-dependent accumulation of cells in G1 phase of
the cell cycle (66.12, 73.88 and 75.67% of cells in G1 phase
following treatment with 0, 5 and 10 µM WIN, respectively), as
compared with vehicle treatment. In addition, treatment with WIN
led to a dose-dependent decrease in the number of cells in S phase
of the cell cycle (26.08, 14.31 and 12.66% cells in S phase
following treatment with 0, 5 and 10 µM WIN, respectively).
As shown in Fig. 1B, WIN inhibited
the proliferation of BEL7402 cells in a dose-dependent manner.
WIN-induced cell cycle arrest is mediated
via the upregulation of p27 and concomitant downregulation of
cyclin D1 and Cdk4
Since the present study demonstrated that treatment
of BEL7402 cells with WIN resulted in a G1 phase arrest, the
effects of WIN on the cell cycle regulatory molecules that operate
in G1 phase of the cell cycle were assessed. The present study also
investigated the role of the Cdk inhibitor (Cki) cyclin Cdk
machinery in the WIN-mediated G1 phase cell cycle arrest of BEL7402
cells. A marked increase in the protein expression of p27 was
detected following treatment with WIN at 5 and 10 µM doses
(Fig. 1C). Using western blot
analysis, the effects of WIN on the protein expression levels of
cyclin D1 and Cdk4, which are known to be regulated by p27, were
investigated. Treatment of BEL7402 cells with WIN led to a
dose-dependent decrease in the protein expression levels of cyclin
D1 and Cdk4 (Fig. 1C).
WIN downregulates the levels of p-Rb and
E2F1
The present study also detected the effects of WIN
on the protein expression levels of pRb and E2F1. Western blot
analysis revealed that treatment of BEL7402 cells with WIN led to a
significant decrease in the protein expression of pRb (Fig. 1D). Since pRb controls the cell
cycle via binding to and inhibiting the E2F1 transcription factor,
the protein expression of E2F1 was also detected. As shown in
Fig. 1D, treatment of BEL7402
cells with WIN led to a dose-dependent decrease in E2F1
expression.
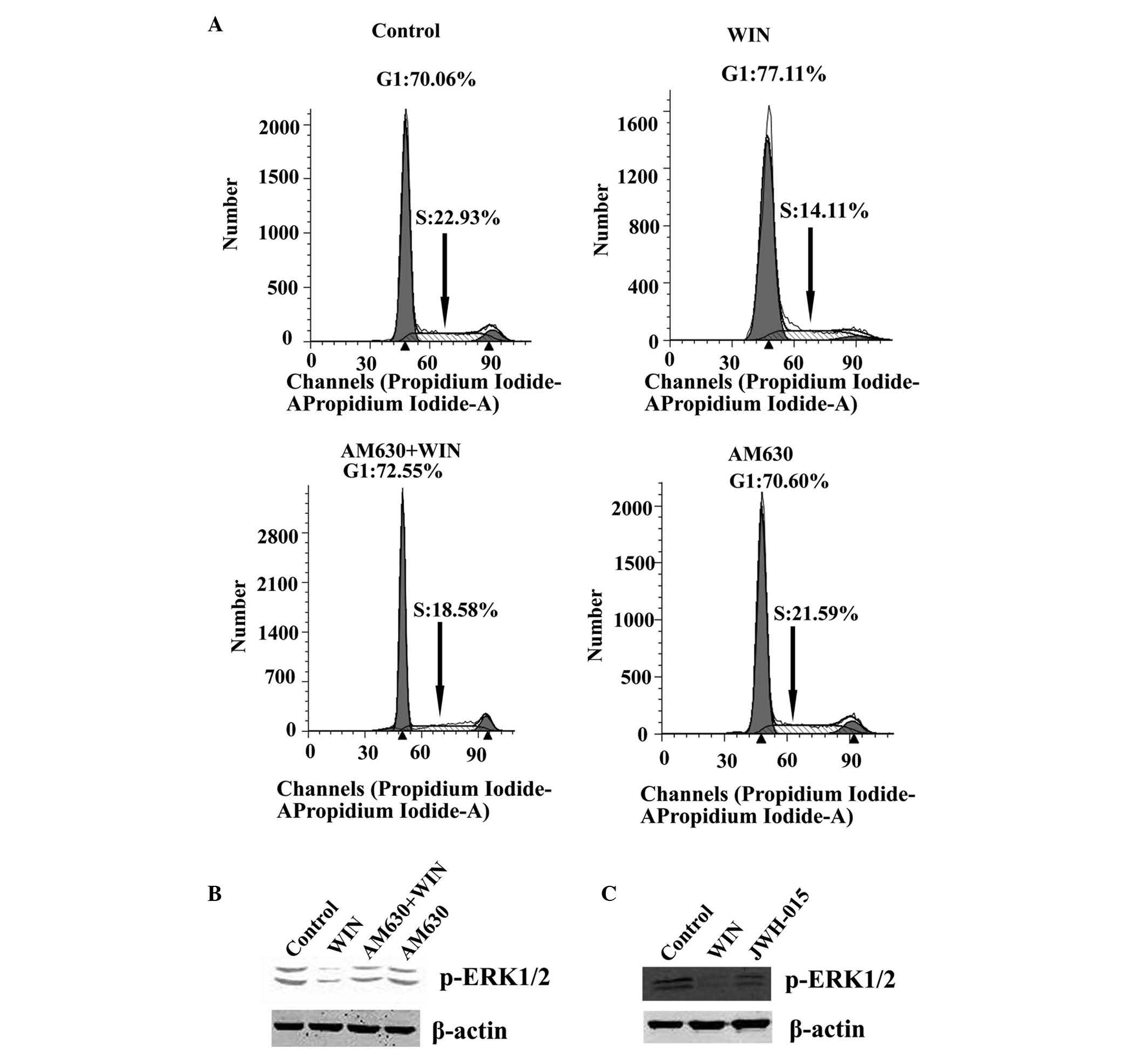
WIN-induced inactivation of ERK results
in cell cycle arrest via CB2
A significant inactivation of ERK1/2 was detected
when the BEL7402 cells were treated with WIN at a dose of 5 and 10
µM (Fig. 1E). To confirm
that ERK1/2 inactivation was cannabinoid receptor-mediated, the
BEL7401 cells were pretreated with the CB2 antagonist, AM630,
followed by treatment with both AM630 and WIN. As shown in Fig. 2B, no alteration in the activation
of ERK1/2 was observed when the cells were treated with the
antagonist alone, as compared with the control group. However,
treatment with WIN led to a marked inactivation of ERK1/2. When the
antagonist was co-administered with WIN, an increase in the protein
expression of p-ERK1/2 was detected, as compared with in the cells
treated with WIN alone. As shown in Fig. 2A, when the cells were treated with
the antagonist alone, no change in the number of cells in G1 phase
(70.60%) and S phase (21.59%) was observed, as compared with the
control group (G1 phase, 70.06%; S phase, 22.93%). Treatment with
WIN led to a significant inhibition in cell cycle progression (G1
phase, 77.11%; S phase, 14.11%). However, when the antagonist was
co-administered with WIN, the inhibition in cell cycle progression
was attenuated (G1 phase, 72.55%; S phase, 18.58%). Furthermore, in
the BEL7402 cells treated with the CB2 selective agonist, JWH-015,
the protein expression of p-ERK1/2 was significantly decreased
(Fig. 2C).
WIN-induced inactivation of ERK1/2
results in cell growth inhibition and cell cycle arrest
To confirm the role of p-ERK1/2 in cannabinoid
receptor-induced cell growth inhibition, the BEL7402 cells were
treated with 30 µM ERK1/2 inhibitor, PD98059, for 24 h.
Treatment with PD98059 alone resulted in a decrease in the
viability of BEL7402 cells in a dose-dependent manner; however,
85.4±0.9% cells survived following treatment with 40
µm PD98059
(Fig. 3A). The present study
demonstrated that treatment of BEL7402 cells with WIN resulted in
G1 phase cell cycle arrest. To determine whether the cell cycle
arrest was mediated by inactivation of ERK1/2, a DNA cell cycle
analysis was performed. As shown in Fig. 3B, inhibiting the activation of
ERK1/2 using the inhibitor PD98059 resulted in an increase in the
number of cells in G1 phase of the cell cycle (75.22%), as compared
with the control group (71.32%). In addition, the number of cells
in S phase of cell cycle was decreased (18.72%), as compared with
the control group (21.40%). Treatment with WIN led to an
accumulation of cells in G1 phase (78.83%) and a reduction of cells
in S phase (10.21%). In addition, the present study determined the
effects of ERK1/2 on the expression of p27, a cell cycle regulatory
molecule that operates in G1 phase of the cell cycle, and cyclin
D1, which is associated with cell proliferation. Treatment with WIN
increased the expression of p27, and treatment with PD98059 also
increased the expression of p27 (Fig.
3C). Furthermore, treatment with WIN or PD98059 markedly
inhibited the expression of cyclin D1 in the BEL7402 cells
(Fig. 3C).
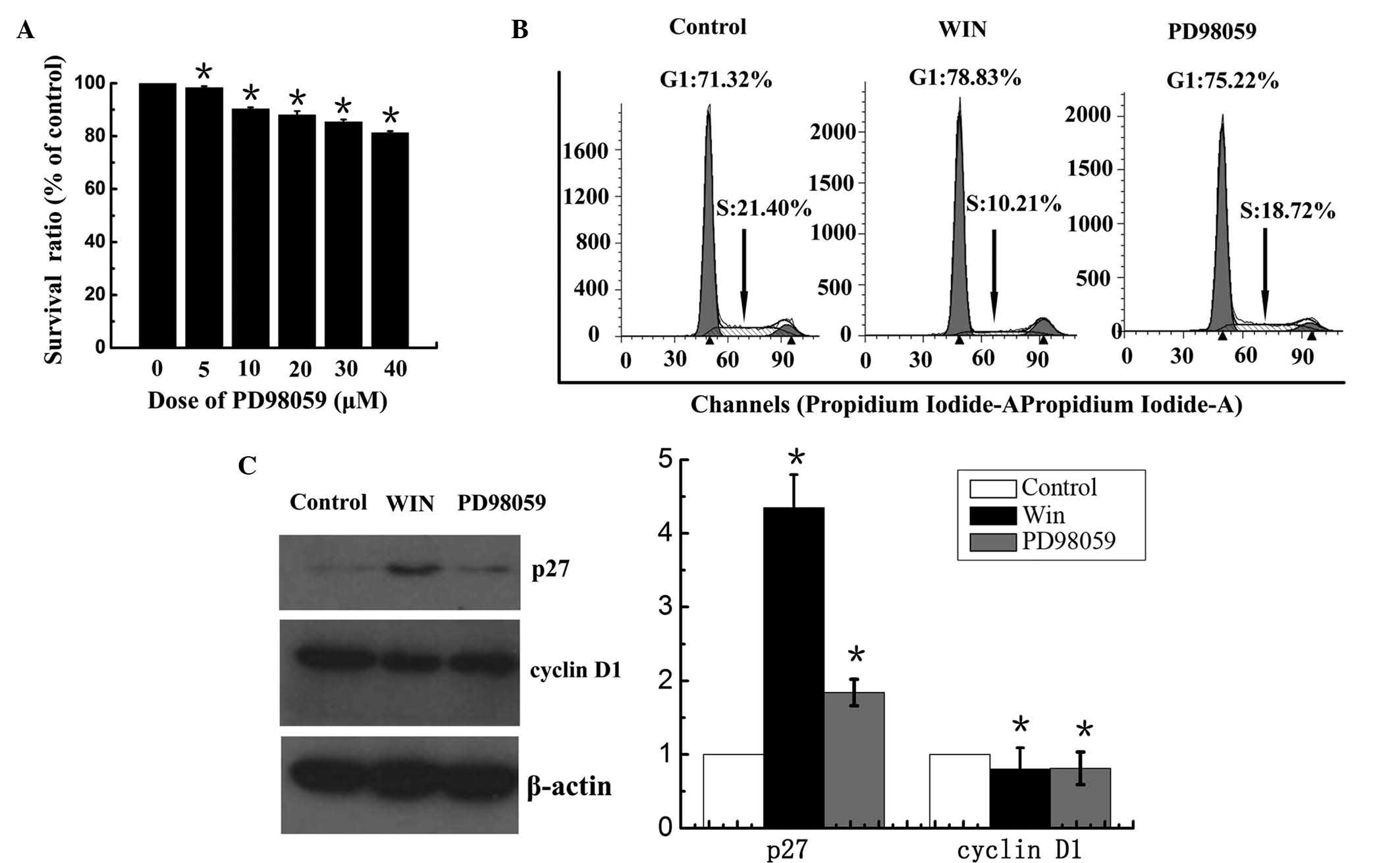 | Figure 3Effects of WIN and ERK1/2 inhibitor on
cell cycle distribution, and cyclin D1 and p27 expression in
BEL7402 hepatocellular carcinoma cells. (A) Effects of ERK1/2
inhibitor, PD98059, on the cell viability of BEL2407 cells. (B)
Effects of the ERK1/2 inhibitor, PD98059, on the cell cycle
distribution of BEL7402 cells. Cell cycle analysis was performed by
flow cytometry and the labeled cells were analyzed using a FACScan
flow cytometer, and the percentage of cells in the G0/G1, S and
G2/M phases was calculated using ModFit LT software. The data are
representative of a typical experiment repeated three times. (C)
Effects of ERK1/2 inhcell lysates were prepared for immunoblot
analysis. The blot shown is representative of a typical experiment
repeated three times. Expression levels were quantified by
densitometric analysis with normalization to β-actin. Values are
expressed as the mean ± standard deviation. *P<0.05
vs. untreated cells. WIN, WIN55, 212-2; ERK, extracellular
signal-regulated kinase. |
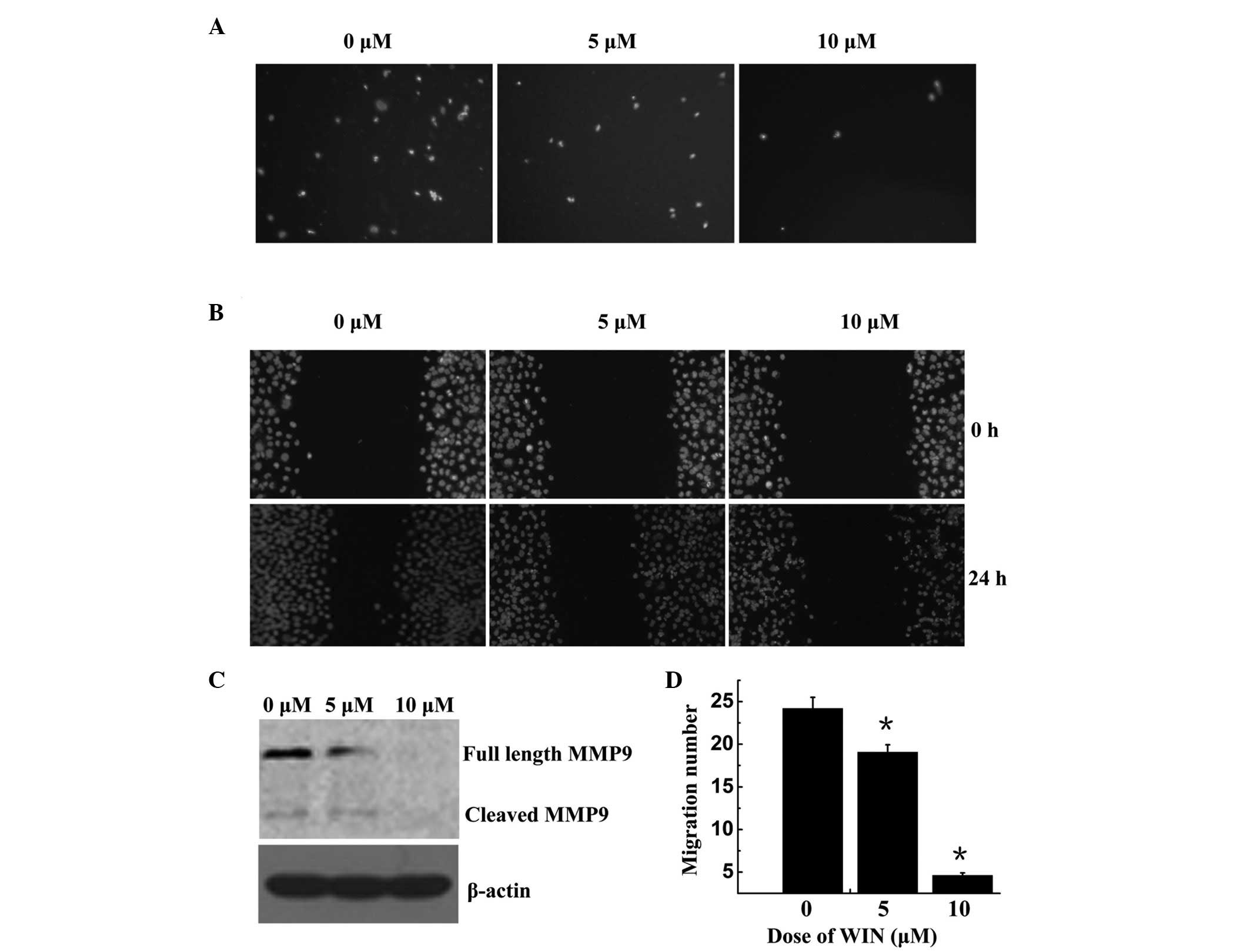
WIN inhibits BEL7402 cell migration via
MMP-9 down-regulation
The present study performed Trans well and wound
healing assays using BEL7402 cells. As shown in Fig. 4A and D, cell migration was
significantly decreased following treatment with WIN in a
dose-dependent manner; the number of migrated cells was 24.20±1.31,
19.07±0.88, and 460±0.29 following treatment with 0, 5 and 10
µM WIN, respectively. The wound healing assay demonstrated
that BEL7402 cells treated with WIN invaded the wound more slowly
compared with the control group, in a dose-dependent manner
(Fig. 4B). MMPs may be associated
with the impaired migration of WIN-treated cells. To explore this
hypothesis, the present study examined the protein expression of M
M P-9. As shown in Fig. 4C, MMP-9
was significantly reduced in the cells treated with WIN, as
compared with the control group.
Discussion
Cannabinoids and their derivatives have recently
attracted attention in the treatment of cancer due to their diverse
abilities, including anti-inflammation, cell growth inhibition and
antitumor properties (6, 10). Previous studies have suggested that
cannabinoid receptors may be an essential target for the treatment
of cancer (14, 15). Our previous study indicated that
WIN-induced apoptosis of BEL7402 HCC cells may be mediated via the
CB2 receptor; therefore, this receptor may be considered as a
potential target for the treatment of HCC (12). A previous study suggested that
cannabinoids inhibit non-small cell lung cancer growth and
metastasis; however, little is currently known regarding the
effects and underlying mechanisms of cannabinoids on non-small cell
lung cancer proliferation and migration (13). The present study aimed to determine
the mechanism underlying the antiproliferative and antimigratory
effects of the cannabinoid agonist, WIN, against HCC. The results
of the present study demonstrated for the first time, to the best
of our knowledge, that the treatment of BEL7402 cells with WIN was
able to inactivate ERK1/2, resulting in cell cycle dysregulation
and G1 arrest. In addition, treatment of BEL7402 cells with WIN
decreased the expression levels of MMP-9, leading to the inhibition
of migration.
ERK1/2 has a dual function and is involved in cell
cycle arrest and proliferation. Inactivation of ERK1/2, and cell
death and proliferation depend on numerous factors. A previous
study demonstrated that cell cycle progression was mediated by the
ERK1/2 and p27 pathway in prostate cancer (11). In addition, treatment with the
ERK1/2 inhibitor, PD98059, downregulated the protein expression
levels of p27 and cyclin D1 in BEL7402 cells, and led to G1 phase
cell cycle arrest. These data suggested that the ERK1/2 pathway may
be involved in the WIN-mediated cell cycle arrest in BEL7402 cells.
Although certain cannabinoids are able to function via transiently
activated vanilloid receptors or lipid rafts, the majority of
cannabinoids act predominantly via cannabinoid receptors (16). The expression levels of CB2 have
previously been shown to be higher in BEL7402 cells, as compared
with in LO2 normal human hepatocytes (12). The present study demonstrated that
WIN significantly down-regulated the expression levels of ERK1/2 in
BEL7402 cells; however, treatment with the CB2 antagonist AM630,
was able to attenuate the inhibition. Treatment with WIN resulted
in a marked accumulation of BEL7402 cells in G1 phase; however,
treatment with the CB2 antagonist AM630, attenuated this effect.
Furthermore, the CB2 selective agonist, JWH-015, decreased the
protein expression levels of ERK1/2 in BEL7402 cells. These data
suggested that cell cycle dysregulation in BEL7402 cells following
treatment with WIN was regulated via CB2, and the p-ERK1/2 and p27
pathway.
It is well established that uncontrolled cellular
growth and metastatic reoccurrence are responsible for the
development of the majority of cancer types, including HCC.
Therefore, agents that can modulate cell cycle and metastasis may
be useful in the management and treatment of cancer. Consistent
with this notion, developing novel targets and mechanism-based
anti-proliferative and anti-migratory agents for the management of
HCC is essential. One of the most promising areas of recent
cannabinoid research is the ability to control cell growth
(8). Numerous studies have
demonstrated that the cannabinoid-mediated inhibition of growth may
be cell cycle-dependent (11,
17). Therefore, the present study
analyzed the effects of WIN on the proportion of cells in various
phases of the cell cycle. WIN was able to induce a dose-dependent
accumulation of cells in G1 phase of the cell cycle, and also
resulted in an inhibition of cell proliferation. Inhibition of the
cell cycle has previously been suggested as a target for the
management of cancer (11). The
present study also investigated the role of the Cki-cyclin-Cdk
machinery in the WIN-mediated G1 phase cell cycle arrest of BEL7402
cells. The eukaryotic cell cycle is regulated by protein kinase
complexes, which are comprised of cyclins (the regulatory subunit),
which bind to Cdks (catalytic subunit), in order to form active
cyclin-Cdk complexes. Cdk activity is additionally regulated by
small proteins, known as Ckis, which include p27. It has been
reported that Ckis inhibit the kinase activities associated with
cyclin-Cdk complexes; therefore, modulating the phosphorylation
events that have a critical role in the progression of the cell
cycle (18). A previous study
demonstrated that cell cycle progression through the G0/G1 phase is
regulated by p27 (19). The
present study revealed that treatment of BEL7402 cells with WIN led
to an increase in the expression levels of p27 and a decrease in
the expression levels of cyclin D1 and Cdk4. These results
indicated that cell cycle dysregulation in BEL7402 cells following
treatment with WIN may be regulated via the p27, cyclin D1 and Cdk4
pathway.
It has previously been reported that downregulation
of Cdk4 results in phosphorylation and inactivation of pRb, which
can in turn downregulate members of the E2F family and inhibit the
transcription of genes required for S phase progression (20). Progression of S phase in the cell
cycle is accomplished by tran-scriptional activation of E2F target
genes via phosphorylation of pocket proteins by Cdks (18). E2F1 is able to regulate the
expression of MMP-9, and therefore induce migration (21–23).
MMPs are involved in cell migration and are frequently upregulated
in cancer cells (24). The present
study demonstrated that treatment of BEL7402 cells with WIN
downregulated the expression levels of pRb and E2F1. Protein
expression levels of MMP-9 were downregulated in BEL7402 cells
following treatment with WIN, which significantly impaired their
migratory capability. Furthermore, Transwell and wound healing
assays indicated that treatment of BEL7402 cells with WIN
significantly inhibited the metastatic capability of the cells.
These results indicated that metastatic inhibition of BEL7402 cells
by WIN may be regulated via the Cdk4, pRb, E2F1 and MMP-9
pathway.
In conclusion, the results of the present study
suggested that WIN may inhibit metastasis via the MMP-9 pathway and
induce cell cycle arrest via ERK1/2 inactivation in BEL7402 HCC
cells. These results provided a basis for the application of WIN in
the treatment of HCC.
The present study was supported by the National
Science Foundation of China (no. 81372466), International
Conference on Transcriptomics, the 2014 undergraduate and graduate
training innovation project of Guangdong province
(2014JGXM-MS20,2014JGXM-MS20) and the project of Guangzhou
Municipality Bureau of Education (no. 11A033).
References
|
1
|
Lin H, van den Esschert J, Liu C and van
Gulik TM: Systematic review of hepatocellular adenoma in China and
other regions. J Gastroenterol Hepatol. 26:28–35. 2011. View Article : Google Scholar
|
|
2
|
Tanaka M, Katayama F, Kato H, Tanaka H,
Wang J, Qiao YL and Inoue M: Hepatitis B and C virus infection and
hepatocellular carcinoma in China: A review of epidemiology and
control measures. J Epidemiol. 21:401–416. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sia D and Villanueva A: Signaling pathways
in hepatocellular carcinoma. Oncology. 81(Suppl 1): 18–23. 2011.
View Article : Google Scholar
|
|
4
|
Qin LX: Recent progress in predictive
biomarkers for metastatic reccurrence of human hepatocellular
carcinoma: A review of the literature. J Cancer Res Clin Oncol.
130:497–513. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tabrizian P, Franssen B, Jibara G, Sweeney
R, Sarpel U, Schwartz M and Labow D: Cytoreductive surgery with or
without hyperthermic intraperitoneal chemotherapy in patients with
peritoneal hepatocellular carcinoma. J Surg Oncol. 110:786–790.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hermanson DJ and Marnett LJ: Cannabinoids,
endocan-nabinoids, and cancer. Cancer Metastasis Rev. 30:599–612.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bowles DW, O'Bryant CL, Camidge DR and
Jimeno A: The intersection between cannabis and cancer in the
United States. Crit Rev Oncol Hematol. 83:1–10. 2012. View Article : Google Scholar
|
|
8
|
Giuliano M, Pellerito O, Portanova P,
Calvaruso G, Santulli A, De Blasio A, Vento R and Tesoriere G:
Apoptosis induced in HepG2 cells by the synthentic cannabinoid WIN:
Involvement of the transcription factor PPARgamma. Biochimie.
91:457–465. 2009. View Article : Google Scholar
|
|
9
|
Calvaruso G, Pellerito O, Notaro A and
Giuliano M: Cannabinoid-associated cell death mechanisms in tumor
models (review). Int J Oncol. 41:407–413. 2012.PubMed/NCBI
|
|
10
|
Chakravarti B, Ravi J and Ganju RK:
Cannabinoids as therapeutic agents in cancer: Current status and
future implications. Oncotarget. 5:5852–5872. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sarfaraz S, Afaq F, Adhami VM, Malik A and
Mukhtar H: Cannabinoid receptor agonist-induced apoptosis of human
prostate cancer cells LNCaP proceeds through sustained activation
of ERK1/2 leading to G1 cell cycle arrest. J Biol Chem.
281:39480–39491. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hong Y, Zhou Y, Wang Y, Xiao S, Liao DJ
and Zhao Q: PPARγ mediates the effects of WIN55,212-2, an synthetic
cannabinoid, on the proliferation and apoptosis of the BEL-7402
hepatocarcinoma cells. Mol Biol Rep. 40:6287–6293. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Preet A, Qamri Z, Nasser MW, Prasad A,
Shilo K, Zou X, Groopman JE and Ganju RK: Cannabinoid receptors,
CB1 and CB2, as novel targets for inhibition of non-small cell lung
cancer growth and metastasis. Cancer Prev Res (Phila). 4:65–75.
2011. View Article : Google Scholar
|
|
14
|
Xian XS, Park H, Choi MG and Park JM:
Cannabinoid receptor agonist as an alternative drug in
5-fuorouracil-resistant gastric cancer cells. Anticancer Res.
33:2541–2547. 2013.PubMed/NCBI
|
|
15
|
Brandi J, Dando I, Palmieri M, Donadelli M
and Cecconi D: Comparative proteomic and phosphoproteomic profiling
of pancreatic adenocarcinoma cells treated with CB1 or CB2
agonists. Electrophoresis. 34:1359–1368. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dainese E, Oddi S, Bari M and Maccarrone
M: Modulation of the endocannabinoid system by lipid rafts. Curr
Med Chem. 14:2702–2715. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zeni O, Sannino A, Romeo S, Micciulla F,
Bellucci S and Scarf MR: Growth inhibition, cell-cycle alteration
and apoptosis in stimulated human peripheral blood lymphocytes by
multi-walled carbon nanotube buckypaper. Nanomedicine (Lond).
10:351–360. 2015. View Article : Google Scholar
|
|
18
|
Sánchez I and Dynlacht BD: New insights
into cyclins, CDKs, and cell cycle control. Semin Cell Dev Biol.
16:311–321. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kumar P and Wood C: Kaposi's
sarcoma-associated herpesvirus transactivator Rta induces cell
cycle arrest in G0/G1 phase by stabilizing and promoting nuclear
localization of p27kip. J Virol. 87:13226–13238. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Deshpande A, Sicinski P and Hinds PW:
Cyclins and cdks in development and cancer: A perspective.
Oncogene. 24:2909–2915. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ma X, Gao Y, Fan Y, Ni D, Zhang Y, Chen W,
Zhang P, Song E, Huang Q, Ai Q, et al: Overexpression of E2F1
promotes tumor malignancy and correlates with TNM stages in clear
cell renal cell carcinoma. PLoS One. 8:e734362013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Johnson JL, Pillai S, Pernazza D, Sebti
SM, Lawrence NJ and Chellappan SP: Regulation of matrix
metalloproteinase genes by E2F transcription factors: Rb-Raf-1
interaction as a novel target for metastatic disease. Cancer Res.
72:516–526. 2012. View Article : Google Scholar :
|
|
23
|
Pillai S, Kovacs M and Chellappan S:
Regulation of vascular endothelial growth factor receptors by Rb
and E2F1: Role of acetylation. Cancer Res. 70:4931–4940. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lukaszewicz-Zając M, Mroczko B and
Szmitkowski M: Gastric cancer - The role of matrix
metalloproteinases in tumor progression. Clin Chim Acta.
412:1725–1730. 2011. View Article : Google Scholar
|