Introduction
Doxorubicin (DOX) is an anthracycline antibiotic,
which is used to treat various types of neoplastic disease in
humans (1). However, the use of
DOX clinically is limited by severe toxic side-effects on the
heart, which can lead to dilated cardiomyopathy and congestive
heart failure (2). Several studies
have shown that the production of reactive oxygen species (ROS) is
implicated in DOX cardiotoxicity, which eventually leads to
endothelial dysfunction (3,4) and
cardiomyocyte apoptosis (5). A
number of pharmacological interventions have been suggested as
therapies to inhibit DOX-induced cardio-toxicity (6–8).
Hydrogen sulfide (H2S) has long bee
considered a toxic gas, however, it has now been qualified as the
third gasotransmitter, along with nitric oxide and carbon monoxide,
exerting various effects in the cardiovascular system (9–11).
H2S has been shown to protect the heart from myocardial
ischemia-reperfusion injury in various studies (12,13).
Our previous study demonstrated that increased endogenous
H2S generation in the early reperfusion phase is
important in ischemia preconditioning (IPC)-elicited protection in
isolated hearts (11).
Various antioxidant signaling pathways protect or
regulate the response of cells to oxidative stress (14,15).
Several antioxidants, including peroxiredoxins (Prxs), are oxidized
in cells treated with ROS. Prxs are a recently characterized family
of antioxidant enzymes, which control the expression levels of
cytokine-induced peroxide and mediate signal transduction in
mammalian cells (16). Multiple
mammalian Prxs (I–VI) often coexist in the same cell in various
intracellular locations and function as scavengers of cellular
H2O2, which are released following
stimulation with growth factors during proliferation, apoptosis or
oxidative stress (17,18). Furthermore, the expression levels
of Prxs are high in the heart and there has been increasing
interest in their importance in the cardiac response to oxidative
stress (19). Notably, Prx III has
a protective role in cisplatin- and gentamicin-induced apoptosis
through a mitochondria-dependent signaling pathway (20). The over-expression of Prx III
protects the mouse myocardium from infarction (21). By contrast, the depletion of Prx
III results in increased intracellular expression levels of
H2O2 and sensitizes cells to apoptotic
signaling (22).
In the present study, H9c2 cells were treated with 5
µM DOX to establish a chemotherapy-induced cardiotoxicity
model (6). This model was then
used to investigate whether DOX induces the expression of Prx III
in the H9c2 cells, and to examine the role of Prx III in the
protective effect of H2S against DOX-induced injury in
H9c2 cells.
Materials and methods
Materials
Methyl thiazolyl tetrazolium (MTT), Hoechst 33258,
DOX, sodium hydrosulfide (NaHS) and N-acetyl-L-cysteine (NAC) were
purchased from Sigma-Aldrich (St. Louis, MO, USA). All cell culture
medium components were purchased from Thermo Fisher Scientific,
Inc. (Waltham, MA, USA), unless otherwise specified. The H9c2
cardiomyocytes were purchased from the Shanghai Cell Library of
China, which were originally obtained from the American Type
Culture Collection (Manassas, VA, USA).
Cell culture
The H9c2 cardiomyocytes were cultured in Dulbecco's
modified Eagle's medium (DMEM; Beyotime Institute of Biotechnology,
Shanghai, China) supplemented with 10% fetal bovine serum (FBS;
Beyotime Institute of Biotechnology), 100 µg/ml streptomycin
(Gibco; Thermo Fisher Scientific, Inc.) and 100 U/ml penicillin
(Gibco; Thermo Fisher Scientific, Inc.) in a humidified atmosphere
containing 5% CO2 at 37°C, and the cells were passaged
every 2 days. The H9c2 cardiomyocytes were seeded at a density of
2×106 cells/dish into 100 mm dishes containing 10% FBS,
and incubated at 37°C for 24 h, following which the medium was
replaced with 0.5% DMEM supplemented with FBS for 24 h
starvation.
MTT assay
The MTT assay, which is a standard method used to
assess the viability of cells, was performed in the present study.
Prior to each experiment, the H9c2 cardiomyocytes (5×103
cells/well) were seeded into 96-well microtitre plates. Following
incubation with NAC for 60 min and/or NaHS for 30 min at 37°C, the
cells were treated with 5 µM DOX for a further 24 h.
Subsequently, 10 µl MTT solution was added to each of the
wells, and the plates were incubated for 4 h at 37°C. The
absorbance was then measured at 470 nm using a SpectraMax 190
spectrophotometer (Molecular Devices, Sunnyvale, CA, USA), and used
to calculate the relative ratio of cell viability. Three
independent experiments were performed for each experimental
condition.
Assessment of cardiomyocyte cell
apoptosis
The levels of apoptosis were analyzed using
fluorescence microscopy following staining of the cells with
Hoechst 33258 chromatin dye. Following the treatments described
above, the cells were fixed with ice-cold 4% paraformaldehyde
dissolved in phosphate-buffered saline (PBS) at room temperature
for 20 min. Non-specific binding was blocked using 5% normal goat
serum (Sigma-Aldrich) in 0.01 M PBS containing 0.3% Triton X-100.
The cells were then washed twice with PBS and incubated with 10
µg/ml Hoechst 33258 for 10 min at room temperature in the
dark. The cells were visualized under a fluorescence microscope
(BX50-FLA; Olympus Corporation, Tokyo, Japan). Apoptotic cells
exhibited condensed, fractured or distorted nuclei, whereas viable
cells exhibited normal nuclear size and uniform fluorescence. The
apoptotic rate was analyzed with a cell counter in Image J 1.4
software (National Institutes of Health, Bethesda, MD, USA).
Western blot analysis
The cells were homogenized directly using cell lysis
buffer (Cell Signaling Technology, Inc., Danvers, MA, USA) and the
lysates were centrifuged at 12,000 × g for 10 min at 4°C. Protein
concentrations were determined with the use of a Bicinchoninic Acid
Protein Assay kit (Beyotime Institute of Biotechnology), according
to the manufacturer's protocol. The extracted proteins (30
µg) were mixed with 5% SDS sample buffer (Beyotime Institute
of Biotechnology) prior to being boiled at 100°C for 7 min and
separated by electrophoresis on a 10% SDS-PAGE. Following
electrophoresis, the proteins were transferred onto polyvinylidene
difluoride membranes (Beyotime Institute of Biotechnology). The
membranes were blocked in Tris-buffered saline with 0.1% Tween 20
(TBS-T), containing 5% non-fat dried milk for 2 h at room
temperature with agitation. Following blocking, the membranes were
incubated with the following antibodies: Rabbit anti-Prx III
polyclonal antibody (cat. no. ab73349; 1:400; Abcam, Cambridge, UK;
1:400, rabbit anti-cystathionine-γ-lyase (CSE) monoclonal antibody
(cat. no. 12217-1-AP; 1:1,000; Proteintech Group, Inc., Chicago,
IL, USA) and GAPDH (cat. no. AG019; dilution 1:1,000; Beyotime
Institute of Biotechnology (Shanghai, China). Subsequently, the
membranes were incubated with 5% milk or bovine serum albumin
overnight at 4°C. Primary antibody was removed by washing the
membranes three times in TBS-T, and incubated for 2 h with the
appropriate horseradish peroxidase-conjugated secondary antibody
(cat. no. A0208; 1:1,000; Beyotime Institute of Biotechnology).
Following three washes in TBS-T, the antigen-antibody bands were
detected using an enhanced chemiluminescence kit (Beyotime
Institute of Biotechnology) and quantified using a densitometry
software program (Quantity One; Bio-Rad Laboratories, Hemel
Hempstead, UK) and GAPDH was used as a loading control.
Statistical analysis
The results of the present study are presented as
the mean ± standard error of the mean. Statistical analysis was
performed using Student's t-test or one-way analysis of variance
with SPSS 13.0 (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
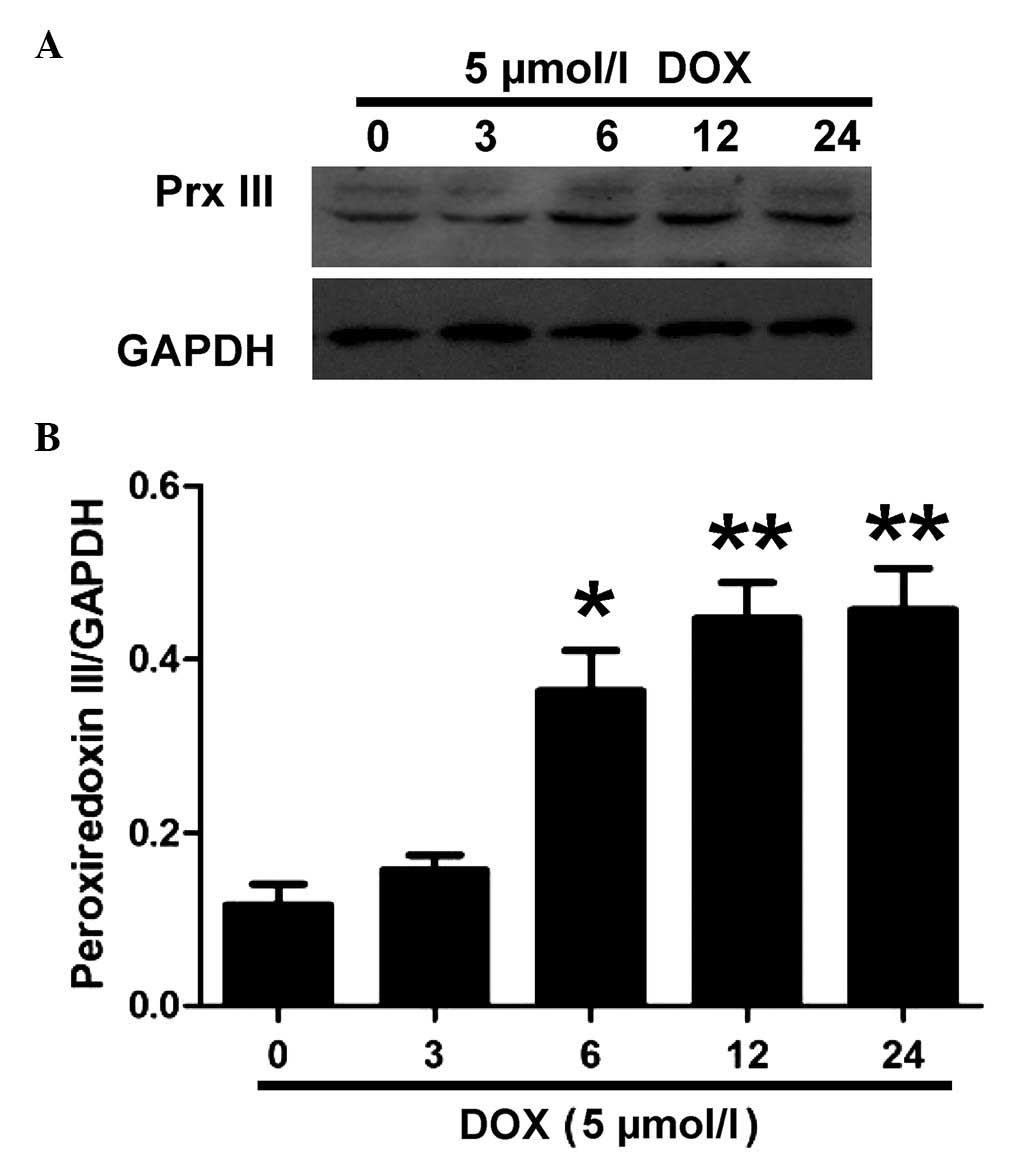
DOX increases the expression levels of
Prx III in H9c2 cells. in a time-dependent manner
To examine the effect of DOX on the expression
levels of Prx III, the H9c2 cells were treated with 5 µM DOX
for the indicated time periods (0, 3, 6, 12 and 24 h). The results
of the western blot analysis demonstrated that DOX increased the
expression levels in of Prx III a time-dependent manner (Fig. 1).
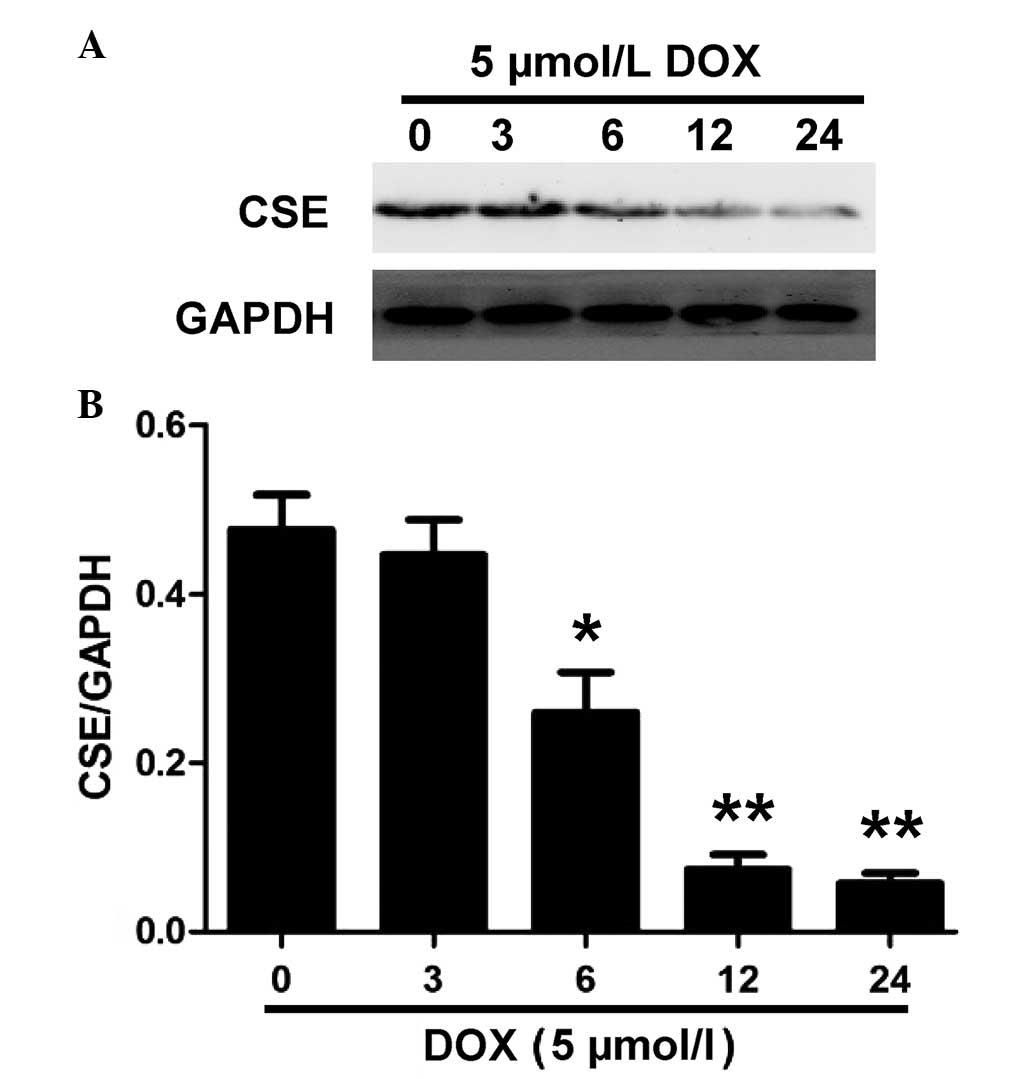
DOX inhibits the expression and activity
of CSE in H9c2 cells
CSE is the enzyme responsible for endogenous
H2S generation in H9c2 cells (23). Western blot analysis was performed
to evaluate whether DOX decreases endogenous H2S
production by inhibiting the expression of CSE. As shown in
Fig. 2, treatment with 5 µM
DOX for the indicated time periods (0, 3, 6, 12 and 24 h) caused a
significant downregulation in the expression of CSE in the H9c2
cells. These results suggested that DOX induced CSE inhibition in
the H9c2 cells, which contributed to a decrease in endogenous
H2S production.
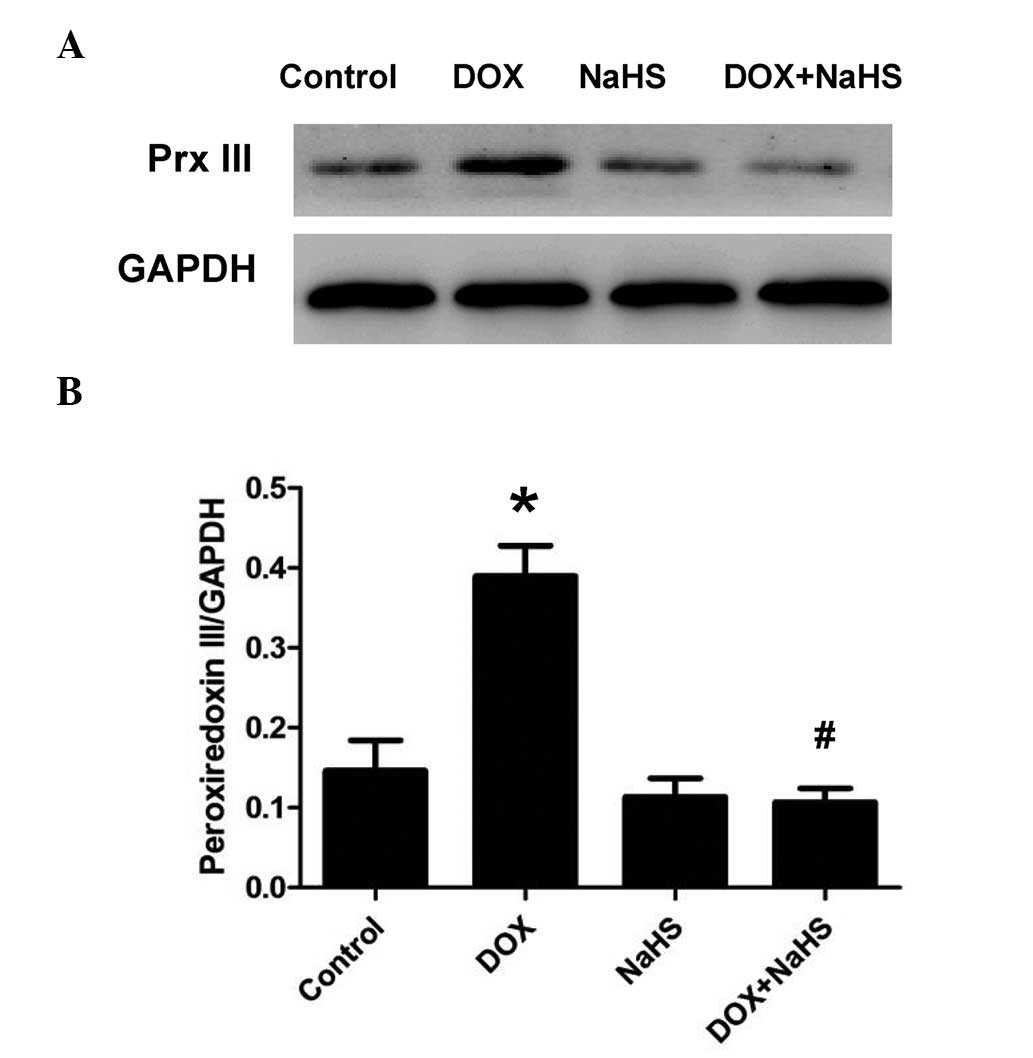
Exogenous H2S inhibits
DOX-induced expression of Prx III in H9c2 cells
To determine whether the protective effect of
H2S against the toxicity induced by DOX was associated
with the inhibition of Prx III, the effects of NaHS on the
expression levels of Prx III induced by DOX were evaluated.
Pretreatment of the H9c2 cells with 100 µmol/l NaHS (a donor
of H2S) for 30 min prior to exposure to 5 µmol/l
DOX for 24 h significantly inhibited the DOX-induced overexpression
of Prx III (Fig. 3). NaHS did not
affect the basal expression levels of Prx III in the H9c2 cells
when treated alone. These results suggested that the inhibition of
Prx III may be involved in the cryoprotective effects of
H2S from DOX-induced cytotoxicity in H9c2 cells.
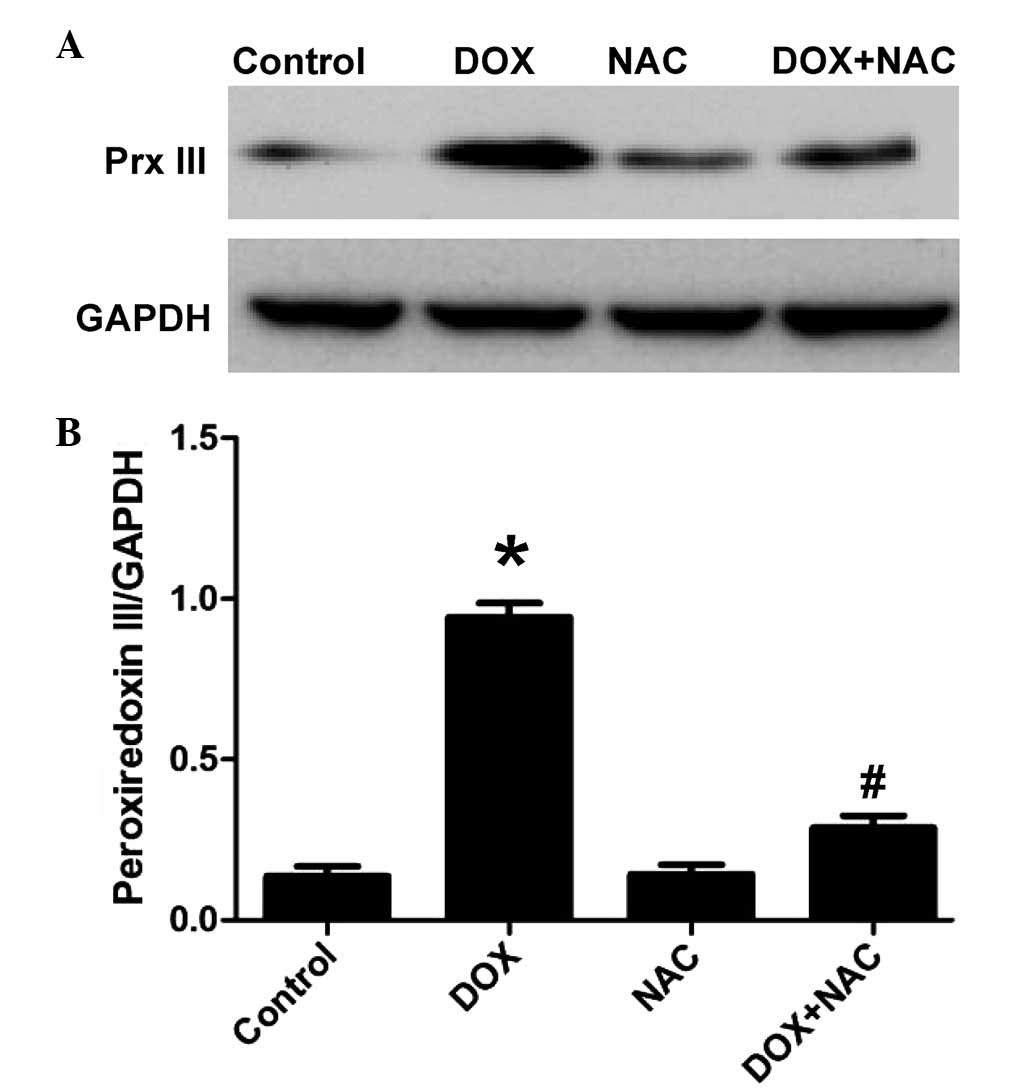
NAC reverses the DOX-induced decrease of
Prx III in H9c2 cells
To determine whether the inhibitory effect of NaHS
on the DOX-induced increased expression of Prx III was associated
with its antioxidant properties, the H9c2 cells were pretreated
with 1,000 µM NAC, a ROS scavenger, for 60 min, following
which the cells were exposed to 5 µM DOX for 24 h. As shown
in Fig. 4, similar to the
inhibitory effect of pretreatment of cells with NaHS, pretreatment
of the H9c2 cells with NAC for 60 min significantly decreased the
expression levels of Prx III. These results suggested that the
inhibitory properties of H2S on the DOX-induced
expression of Prx III involved the contribution of an antioxidant
effect.
Exogenous H2S inhibits
DOX-induced cytotoxicity
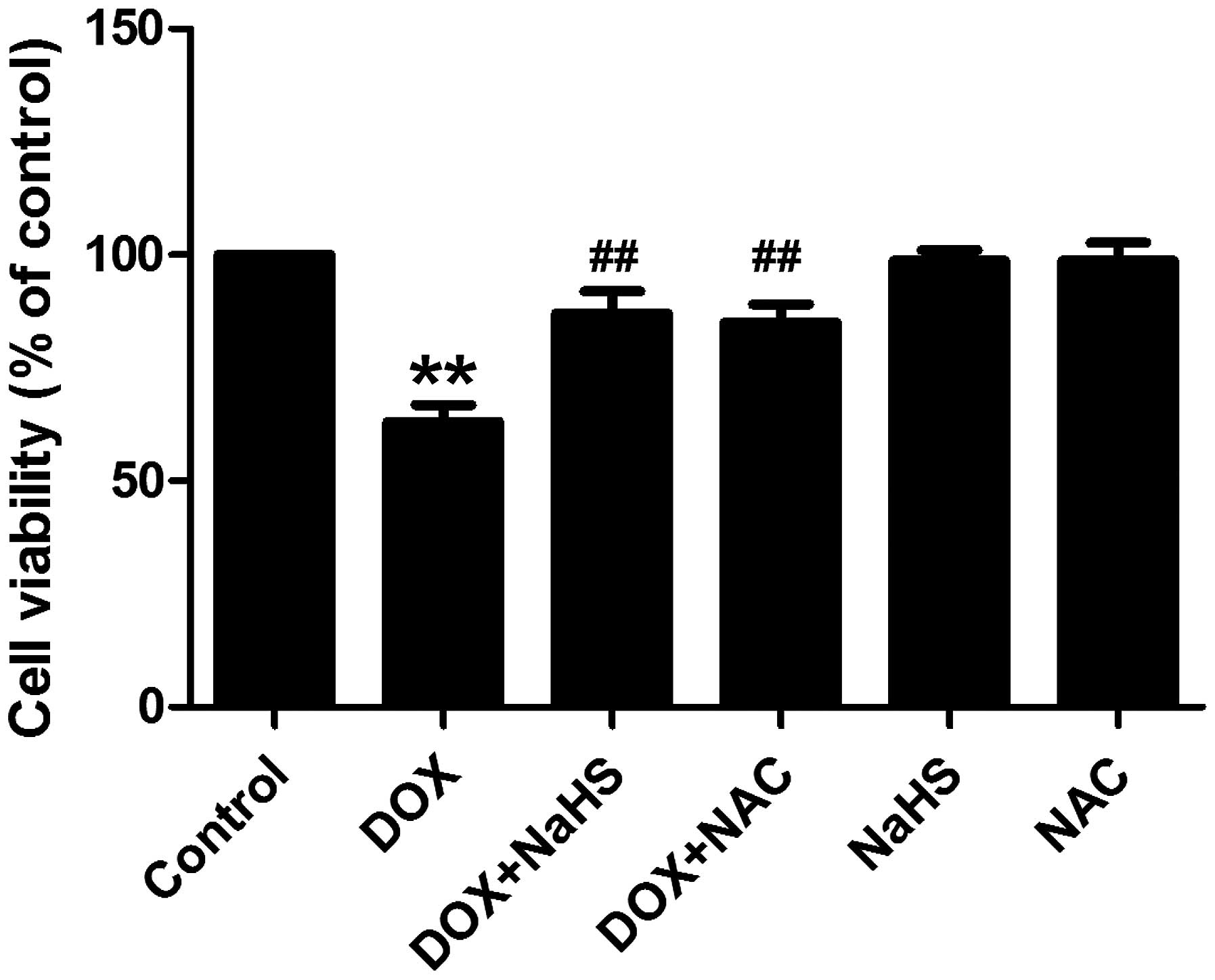
As shown in Fig. 5,
exposure of the H9c2 cells to DOX at 5 µM for 24 h induced
significant cytotoxicity, resulting in a reduction in the viability
of the cells. However, pretreatment of the cells with 100 µM
NaHS for 30 min prior to exposure to DOX significantly reduced the
effects of DOX-induced cytotoxicity, which was demonstrated by an
increase in the viability of the cells. Pretreatment of the cells
with NAC had a similar protective effect as H2S against
DOX-induced cytotoxicity, also suggesting the involvement of an
antioxidant effect against the cytotoxicity induced by DOX.
Treatment with either NaHS or NAC alone did not affect the
viability of the H9c2 cells.
Exogenous H2S reduces
DOX-induced apoptosis in H9c2 cells
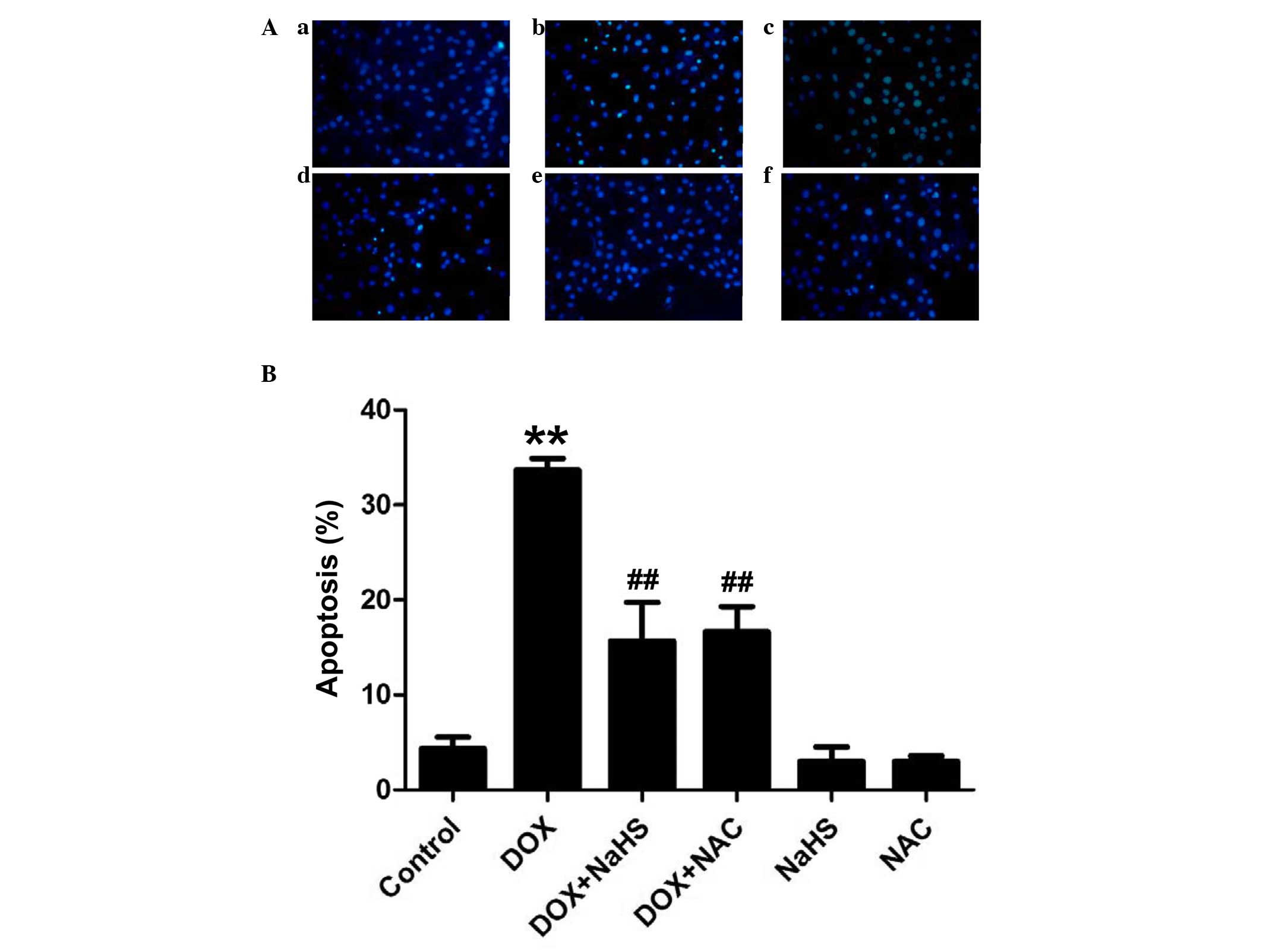
The effects of NaHS and NAC on DOX-induced apoptosis
were further investigated in the present study. As shown in
Fig. 6, the H9c2 cells treated
with 5 µM DOX for 24 h exhibited characteristics typical of
apoptosis, including the condensation of chromatin, nuclear
shrinkage and the presence of apoptotic bodies. However,
pretreatment of the H9c2 cells with 100 µM NaHS for 30 min
prior to DOX exposure significantly decreased the DOX-induced
increase in cells exhibiting nuclear condensation and
fragmentation. In addition, the H9c2 cells were preconditioned with
the ROS scavenger, NAC (1,000 µM) prior to exposure to DOX.
The results demonstrated that pretreatment of the cells with NAC
significantly attenuated DOX-induced H9c2 cell apoptosis. Treatment
with NaHS or NAC alone did not have a marked effect on the
morphology of the cells, or on the number of apoptotic H9c2 cells
identified. These findings suggested that antioxidant properties
are involved in, and contribute to, the inhibitory effects of
H2S on the DOX-induced apoptosis of H9c2 cells.
Discussion
Several studies have demonstrated that the major
molecular mechanism underlying DOX-induced cardiotoxicity are free
radical-induced oxidative stress and cardiomyocyte death caused by
apoptosis and necrosis (6,24). Concordant with previous studies
(24,25), the present study demonstrated that
exposure of H9c2 cells to DOX markedly increased cellular injury,
decreased cell viability, increased cell apoptosis and increased
the expression levels of Prx III.
A previous study demonstrated the cardioprotective
effects of H2S in animal models of disease (26). Treatment with H2S
significantly reduced myocardial infarct size, improved regional
left ventricular function, and increased endothelium-dependent and
endothelium-independent microvascular reactivity in a porcine model
of myocardial ischemia-reperfusion (27). In addition, H2S has been
observed to attenuate myocardial necrosis and apoptosis (28). Endogenous H2S has been
associated with cardioprotection, enabled via metabolic inhibition
preconditioning in rat ventricular myocytes (29). The inhibition of the production of
endogenous H2S by its synthesis inhibitor,
DL-propargylglycine, has been observed to inhibit the protective
effects of IPC in isolated hearts and cardiomyocytes (30). In the present study, H9c2 cells
were used to investigate the effects of DOX on endogenous
H2S generation. The results demonstrated that exposure
of the H9c2 cells to DOX led to a significant decrease in
H2S generation.
Prx III is a mitochondrial antioxidant protein and
member of the Prx family, which is capable of catalyzing
H2O2 reduction (31). In addition, the overexpression of
Prx III has been demonstrated to protect neurons against oxidative
stress-induced cell death (32).
These beneficial effects make Prx III a valuable potential
candidate for the treatment of left ventricular systolic
dysfunction following myocardial infarction, in which the
production of ROS has been observed to be increased in the
mitochondria (21). Although
several previous studies have shown the beneficial effects of
antioxidants in heart failure (33,34),
investigations have not been performed to specifically examine the
role of Prx III in DOX-induced cytotoxicity. The results of the
present study demonstrated that the expression levels of Prx III
were significantly increased in the DOX-induced H9c2 cell injury
model. Similarly, an increase of Prx III was previously reported by
Xi et al (35), and nitrate
treatment restored the expression of Prx III. In the present study,
the results demonstrated that the Prx III expression levels of Prx
III increased following DOX treatment, and exogenous H2S
preconditioning suppressed the expression of Prx III, markedly
attenuating DOX-induced apoptosis.
In previous years, several studies have demonstrated
that ROS are important in the pathogenesis of cardiac failure
(36–38). In addition, antioxidants have been
reported to exert protective effects against heart failure
(21). Oxidative stress is a
primary mechanism by which DOX-induced cardiomyocyte injury
(24). Notably, the present study
demonstrated that oxidative stress was involved in DOX-induced cell
injury, and further examined whether the activation of Prx III by
DOX was due to the induction of ROS. It was shown that
pre-treatment of the H9c2 cells with the NAC ROS scavenger
significantly decreased DOX-induced expression levels of Prx III.
Collectively, these results supported the hypothesis that the
induction of ROS by DOX activates Prx III in H9c2 cells.
In conclusion, the principal finding of the present
study was that H2S inhibited DOX-induced apoptosis in
the H9c2 cells, and its effects may involve inhibition of the
ROS-mediated expression of Prx III. The results of the present
study not only improved current understanding of the mechanisms
underlying H2S-mediated anti-apoptosis in
cardiomyocytes, but they also provide valuable evidence in support
of the potential use of H2S as a treatment for
cardiovascular diseases.
Acknowledgments
The present study was supported by grants from the
Medical Scientific Research Funds of the Guangdong province (grant
no. A2014810) and the Graduate Student Research Innovation Project
of the Hunan province (grant no. CX2013B397).
References
|
1
|
Menna P, Recalcati S, Cairo G and Minotti
G: An introduction to the metabolic determinants of anthracycline
cardiotoxicity. Cardiovasc Toxicol. 7:80–85. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Barry E, Alvarez JA, Scully RE, Miller TL
and Lipshultz SE: Anthracycline-induced cardiotoxicity: Course,
pathophysiology, prevention and management. Expert Opin
Pharmacother. 8:1039–1058. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jang WJ, Choi DY and Jeon IS: Vascular
endothelial dysfunction after anthracyclines treatment in children
with acute lymphoblastic leukemia. Korean J Pediatr. 56:130–134.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Truong J, Yan AT, Cramarossa G and Chan
KK: Chemotherapy-induced cardiotoxicity: Detection, prevention and
management. Can J Cardiol. 30:869–878. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Spagnuolo RD, Recalcati S, Tacchini L and
Cairo G: Role of hypoxia-inducible factors in the
dexrazoxane-mediated protection of cardiomyocytes from
doxorubicin-induced toxicity. Br J Pharmacol. 163:299–312. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guo R, Lin J, Xu W, Shen N, Mo L, Zhang C
and Feng J: Hydrogen sulfide attenuates doxorubicin-induced
cardiotoxicity by inhibition of the p38 MAPK pathway in H9c2 cells.
Int J Mol Med. 31:644–650. 2013.PubMed/NCBI
|
|
7
|
Gu J, Hu W and Zhang DD: Resveratrol, a
polyphenol phytoalexin, protects against doxorubicin-induced
cardiotoxicity. J Cell Mol Med. 19:2324–2328. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Karimi G, Ramezani M and Abdi A:
Protective effects of lycopene and tomato extract against
doxorubicin-induced cardio-toxicity. Phytother Res. 19:912–914.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Polhemus DJ and Lefer DJ: Emergence of
hydrogen sulfide as an endogenous gaseous signaling molecule in
cardiovascular disease. Circ Res. 114:730–737. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang Y, Tang ZH, Ren Z, Qu SL, Liu MH,
Liu LS and Jiang ZS: Hydrogen sulfide, the next potent preventive
and therapeutic agent in aging and age-associated diseases. Mol
Cell Biol. 33:1104–1113. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang YE, Tang ZH, Xie W, Shen XT, Liu MH,
Peng XP, Zhao ZZ, Nie DB, Liu LS and Jiang ZS: Endogenous hydrogen
sulfide mediates the cardioprotection induced by ischemic
post-conditioning in the early reperfusion phase. Exp Ther Med.
4:1117–1123. 2012.PubMed/NCBI
|
|
12
|
Polhemus DJ, Calvert JW, Butler J and
Lefer DJ: The cardioprotective actions of hydrogen sulfide in acute
myocardial infarction and heart failure. Scientifica (Cairo).
2014:7686072014.
|
|
13
|
Li C, Hu M, Wang Y, Lu H, Deng J and Yan
X: Hydrogen sulfide preconditioning protects against myocardial
ischemia/reperfusion injury in rats through inhibition of
endo/sarcoplasmic reticulum stress. Int J Clin Exp Pathol.
8:7740–7751. 2015.PubMed/NCBI
|
|
14
|
Sahu BD, Mahesh Kumar J and Sistla R:
Baicalein, a Bioflavonoid, Prevents Cisplatin-Induced Acute Kidney
Injury by Up-RegulatingAntioxidant Defenses and Down-Regulating the
MAPKs and NF-κB Pathways. PLoS One. 10:e01341392015. View Article : Google Scholar
|
|
15
|
Liu MH, Yuan C, He J, Tan TP, Wu SJ, Fu
HY, Liu J, Yu S, Chen YD, Le QF, Tian W, Hu HJ, Zhang Y and Lin XL:
Resveratrol protects PC12 cells from high glucose-induced
neurotoxicity via PI3K/Akt/FoxO3a pathway. Cell Mol Neurobiol.
35:513–522. 2015. View Article : Google Scholar
|
|
16
|
Wood ZA, Poole LB and Karplus PA:
Peroxiredoxin evolution and the regulation of hydrogen peroxide
signaling. Science. 300:650–653. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Poynton RA and Hampton MB: Peroxiredoxins
as biomarkers of oxidative stress. Biochim Biophys Acta.
1840:906–912. 2014. View Article : Google Scholar
|
|
18
|
Jeong HJ, Jeong HW, Song SS, Kang JW, Seo
JH, Lee YH, Lee KS and Kim DW: Upregulation of peroxiredeoxin III
in the hippocampus of acute immobilization stress model rats and
the Foxo3a-dependent expression in PC12 cells. Cell Mol Neurobiol.
31:1041–1046. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nemoto S, Combs CA, French S, Ahn BH,
Fergusson MM, Balaban RS and Finkel T: The mammalian
longevity-associated gene product p66shc regulates mitochondrial
metabolism. J Biol Chem. 281:10555–10560. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chae HZ, Kim HJ, Kang SW and Rhee SG:
Characterization of three isoforms of mammalian peroxiredoxin that
reduce peroxides in the presence of thioredoxin. Diabetes Res Clin
Pract. 45:101–112. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Matsushima S, Ide T, Yamato M, Matsusaka
H, Hattori F, Ikeuchi M, Kubota T, Sunagawa K, Hasegawa Y, Kurihara
T, et al: Overexpression of mitochondrial peroxiredoxin-3 prevents
left ventricular remodeling and failure after myocardial infarction
in mice. Circulation. 113:1779–1786. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chang TS, Cho CS, Park S, Yu S, Kang SW
and Rhee SG: Peroxiredoxin III, a mitochondrion-specific
peroxidase, regulates apoptotic signaling by mitochondria. J Biol
Chem. 279:41975–41984. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kimura H: Hydrogen sulfide: its
production, release and functions. Amino Acids. 41:113–121. 2011.
View Article : Google Scholar
|
|
24
|
Guo R, Wu K, Chen J, Mo L, Hua X, Zheng D,
Chen P, Chen G, Xu W, Feng J, et al: Exogenous hydrogen sulfide
protects against doxorubicin-induced inflammation and cytotoxicity
by inhibiting p38MAPK/NFkappaB pathway in H9c2 cardiac cells. Cell
Physiol Biochem. 32:1668–1680. 2013.
|
|
25
|
Wang X, Wang XL, Chen HL, Wu D, Chen JX,
Wang XX, Li RL, He JH, Mo L, Cen X, et al: Ghrelin inhibits
doxorubicin cardio-toxicity by inhibiting excessive autophagy
through AMPK and p38-MAPK. Biochem Pharmacol. 88:334–350. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ji Y, Pang QF, Xu G, Wang L, Wang JK and
Zeng YM: Exogenous hydrogen sulfide postconditioning protects
isolated rat hearts against ischemia-reperfusion injury. Eur J
Pharmacol. 587:1–7. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Osipov RM, Robich MP, Feng J, Liu Y,
Clements RT, Glazer HP, Sodha NR, Szabo C, Bianchi C and Sellke FW:
Effect of hydrogen sulfide in a porcine model of myocardial
ischemia-reperfusion: Comparison of different administration
regimens and characterization of the cellular mechanisms of
protection. J Cardiovasc Pharmacol. 54:287–297. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sodha NR, Clements RT, Feng J, Liu Y,
Bianchi C, Horvath EM, Szabo C and Sellke FW: The effects of
therapeutic sulfide on myocardial apoptosis in response to
ischemia-reperfusion injury. Eur J Cardiothorac Surg. 33:906–913.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pan TT, Feng ZN, Lee SW, Moore PK and Bian
JS: Endogenous hydrogen sulfide contributes to the cardioprotection
by metabolic inhibition preconditioning in the rat ventricular
myocytes. J Mol Cell Cardiol. 40:119–130. 2006. View Article : Google Scholar
|
|
30
|
Bian JS, Yong QC, Pan TT, Feng ZN, Ali MY,
Zhou S and Moore PK: Role of hydrogen sulfide in the
cardioprotection caused by ischemic preconditioning in the rat
heart and cardiac myocytes. J Pharmacol Exp Ther. 316:670–678.
2006. View Article : Google Scholar
|
|
31
|
Song IS, Kim HK, Jeong SH, Lee SR, Kim N,
Rhee BD, Ko KS and Han J: Mitochondrial peroxiredoxin III is a
potential target for cancer therapy. Int J Mol Sci. 12:7163–7185.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hattori F, Murayama N, Noshita T and
Oikawa S: Mitochondrial peroxiredoxin-3 protects hippocampal
neurons from excitotoxic injury in vivo. J Neurochem. 86:860–868.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Betge S, Lutz K, Roskos M and Figulla HR:
Oral treatment with probucol in a pharmacological dose has no
beneficial effects on mortality in chronic ischemic heart failure
after large myocardial infarction in rats. Eur J Pharmacol.
558:119–127. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jain AK and Mehra NK: Challenges and
Opportunities. Curr Pharm Des. 21:4441–4455. 2015. View Article : Google Scholar
|
|
35
|
Xi L, Zhu SG, Hobbs DC and Kukreja RC:
Identification of protein targets underlying dietary
nitrate-induced protection against doxorubicin cardiotoxicity. J
Cell Mol Med. 15:2512–2524. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schwarzer M, Osterholt M, Lunkenbein A,
Schrepper A, Amorim P and Doenst T: Mitochondrial reactive oxygen
species production and respiratory complex activity in rats with
pressure overload-induced heart failure. J Physiol. 592:3767–3782.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Münzel T, Gori T, Keaney JF Jr, Maack C
and Daiber A: Pathophysiological role of oxidative stress in
systolic and diastolic heart failure and its therapeutic
implications. Eur Heart J. 36:2555–2564. PubMed/NCBI
|
|
38
|
Zuo L, Chuang CC and Hemmelgarn BT:
Defining the function of ROS and NO. J Appl Physiol. 119:944–951.
2015. View Article : Google Scholar
|