Introduction
Acute respiratory distress syndrome (ARDS) is
frequently associated with trauma, shock and sepsis, and is
characterized by an intense inflammatory process involving the
accumulation of activated neutrophils and the production of
pro-inflammatory cytokines, which causes damage of
alveolar-capillary integrity with subsequent high-permeability and
non-hydrostatic pulmonary edema (1). The vital importance of the pulmonary
microvascular barrier function is demonstrated by the balance
between competing endothelial cell (EC) contractile forces and
adhesive cell-cell and cell-matrix tethering forces, all of which
are closely linked through the endothelial cytoskeleton (2). The tumor necrosis factor α (TNF-α),
interleukin (IL) and platelet-activating factor inflammatory
factors, which induce pulmonary EC cytoskeletal rearrangement,
increase paracellular gap formation and enhance microvascular
permeability underlie the pathognomonic features of ARDS (1).
Ras homolog gene family, member A (RhoA),
ras-related C3 botulinum toxin substrate 1 (Rac1) and cell division
control protein 42 homolog (Cdc42), which belong to the Rho GTPase
family, have been demonstrated to be important in the regulation of
microvascular barrier maintenance by affecting the actin
cytoskeleton (3). Evidence
suggests that Rac and Rho have opposing roles in the regulation of
barrier maintenance and stabilization. Waschke et al
(4) demonstrated that acute
inhibition of the small GTPase Rac1 caused a significant increase
in the permeability of venular microvessels. Furthermore, various
barrier-stabilizing mediators and the barrier-protective cyclic
adenosine monophosphate-regulated, Rho, guanine nucleotide exchange
factor (GEF) exchange protein directly activated by cAMP-1 have
been shown to reduce microvascular permeability, at least in part,
via the activation of Rac1 (5,6). It
is also well-established that Rac1 is able to effectively stabilize
the endothelial barrier via the strengthening of cortical actin,
which can promote the stabilization of junctional proteins
(7). However, unlike other factors
in the microvascular endothelium, for example thrombin, vascular
endothelial growth factor (VEGF) induces the activation of Rac1 and
the rapid production of reactive oxygen species (ROS), causing
endothelial barrier dysfunction (8). Furthermore, previous studies on the
macrovascular endothelium have indicated that the activation of
Rac1 may cause barrier destabilization, possibly through the
generation of ROS (9–11). Therefore, whether Rac1 exerts
protective or detrimental effects on barrier function depends on
the types of cell and various inflammatory conditions.
High mobility group box 1 (HMGB1), originally
identified as an important endogenous signaling molecule, is
released by necrotic and inflammatory cells and has potent
pro-inflammatory properties (12).
HMGB1 induces the expression of adhesion molecules, including
intercellular adhesion molecule 1 and vascular adhesion molecule 1,
and promotes the upregulation of pro-inflammatory cytokines,
including TNF-α, IL-8 and plasminogen activator inhibitor 1, partly
through the interaction of HMGB1 with its receptors (13–16).
HMGB1 is different from early-acting mediators, including TNF-α and
IL-1, and is recognized as a late-acting cytokine with a long
duration of action, which is released with a lag phase of 16–24 h
following endotoxin exposure (12). It is well-documented that HMGB1
contributes to the development of acute lung injury, and the
underlying mechanisms may be associated with activation of the
mitogen-activated protein kinase (MAPK) signaling pathway and
nuclear factor-κB via the interaction of HMGB1 with its receptors
(15,16).
It is noteworthy that, whether Rac1 is involved in
HMGB1-induced hyperpermeability of the pulmonary microvascular
endothelium and its associated molecular mechanisms remain to be
fully elucidated. To investigate these gaps in current knowledge,
the present study investigated pulmonary microvascular endothelial
cells (PMVECs) with recombinant HMGB1, examining the role of HMCB1
and determining the mechanisms underlying the effects of Rac1 in
HMGB1-mediated endothelial barrier function, which may provide
novel therapeutic strategies in the treatment of ARDS via targeting
Rac1.
Materials and methods
Reagents
Recombinant human HMGB1 was purchased from
Sigma-Aldrich (St. Louis, MO, USA). The silencing RNA transfection
reagent, siPORT™ Amine, was purchased from Ambion (Thermo Fisher
Scientific, Waltham, MA, USA). The polyclonal rabbit anti-Rac1
(1:500; sc-217), monoclonal mouse anti-extracellular
signal-regulated kinase (ERK) (1:500; sc-514302) and polyclonal
rabbit anti-p38 (1:500; sc-535) antibodies were purchased from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). The monoclonal
mouse anti-phosphorylated (p-)-ERK (1:500; 9101) and polyclonal
rabbit anti-p-p38 (1:500; 9211) antibodies were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). 8-CPT was purchased
from Sigma-Aldrich. An enhanced chemiluminescence kit was obtained
from Pierce Biotechnology, Inc. (Rockford, IL, USA).
Cell culture
The PMVECs were obtained from American Type Culture
Collection (Manassas, VA, USA) and grown in the Endothelial Growth
Medium-2 (Lonza Group Ltd., Basel, Switzerland) supplemented with
growth factors including VEGF, fibroblast growth factor, insulin
growth factor-1 and epidermal growth factor (Gibco; Thermo Fisher
Scientific), and 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific), according to the manufacturer's protocol. The cells
were grown at 37°C in an atmosphere containing 5% CO2,
and those used for further experimentation were obtained from
passage 6–9. For experimentation, the ECs were plated at an
appropriate density, and used 3 days after plating, unless
otherwise specified. Furthermore, the medium was replaced 1 day
prior to all experiments. During experimentation, the PMVECs
(5×105) were challenged with recombinant human HMGB1 at
different doses (5, 10, 15 and 20 µg/ml) for 2 h. In
addition, 8-CPT (5 µM) was used to promote Rac1 in the
PMVECs.
RNA interference
Dharmacon ON-TARGETplus small interfering RNA
(siRNA) against Rac1 was obtained as pools of four siRNA duplexes
from GE Healthcare Life Sciences, (Chalfont, UK). ON-TARGETplus
siRNA (siControl) targeting a non-human protein, luciferase, was
used as a negative control siRNA with minimal off-target silencing.
The silencing protocol was optimized to allow transfection of the
cells shortly following plating and on non-conventional substrates,
including gold electrodes. At 3–5 h following cell plating, the
siRNA, which was calculated at a final concentration of 100 nM
siRNA, was premixed with transfection reagent (4 µl/ml
siPORT™ Amine, Thermo Fisher Scientific) for 5 min and then diluted
with basal media twice, based on using half the volume typical for
a dish/well. After 16–24 h, an equal volume of serum media was
added to the media containing siRNA. At 48 h post-transfection, the
diluted siRNA media was replaced with serum media (1 ml/plate). The
silenced cells were used 3–6 days post-transfection, and the media
was replaced 1 day prior to all experiments.
Electrical resistance measurements
An electrical cell-substrate impedance sensing
(ECIS) system (Applied Biophysics, Troy, NY, USA) was used to
measure transendothelial electrical resistance (TER), using ECs
grown on gold microelectrodes, as previously described by Wolfson
et al (15). The ECs
(5×105) were plated directly onto the gold
micro-electrodes of the ECIS arrays (8W10E), and cultured for a
minimum of 2 days to establish confluency. Data pooling and
analysis were performed using in-house-created Epool software
(version 2.0), which has integrated graphing associated with
Microsoft Excel (Microsoft, Redmond, WA, USA).
Rac1 activity assay
The activity levels of Rac1 were determined using a
pull-down assay with an Rac Activation Assay kit (Cell Biolabs,
Inc., San Diego, CA, USA), according to the manufacturer's
protocol. Briefly, cells were lysed at 4°C in a pull-down lysis
buffer. p21-activated kinase-conjugated protein beads were
incubated with the cell lysates at 4°C for 1 h. Eluted proteins
were subjected to SDS-PAGE, followed by immunoblotting with the
anti-Rac1 antibody.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
SYBR® Green RT-qPCR (Takara Bio., Inc.,
Otsu, Japan) was used to detect mRNA expression levels. RNA was
extracted from the cells using TRIzol reagent (Invitrogen; Thermo
Fisher Scientific) and was reverse transcribed into cDNA (1
µg) using a Prime Script RT Reagent kit (Takara Bio, Inc.)
under the following conditions: 37°C for 15 min, 85°C for 5 sec and
4°C for 1 min. RT-qPCR was performed with ABI 7500 (Applied
Biosystems; Thermo Fisher Scientific) using SYBR Premix Ex
Taq (Takara Bio, Inc.) under the following thermocycling
conditions: One cycle at 95°C for 30 min; 40 cycles at 95°C for 5
sec and 60°C for 34 sec; one cycle at 95°C for 15 sec, 60°C for 1
min and 95°C for 15 sec. The relative quantification values for
these gene expression levels were calculated using the ΔΔCq
(14) method and normalized using
a house keeping gene. The following primer sequences were used:
Rac1, forward 5′-GACCAGCCGACTAGCTTTTG-3′ and reverse
5′-CGAAGGGATGCTCAAGAGAC-3′; GAPDH, forward
5′-AGGTCGGTGTGAACGGATTTG-3′ and reverse 5′-GGGGTCGTTGATGGCAACA-3′
(Shanghai Shengong Biotechnology Co., Ltd., Shanghai, China). For
cycling the ABI 7500 Real-Time PCR system (Thermo Fisher
Scientific) was used.
Western blotting
The cells were washed once with cold endothelial
basal medium (Lonza Group Ltd., Basel, Switzerland) and total
protein was extracted using 0.3% SDS (Sigma-Aldrich) in 10 mM Tris
lysis buffer (300 µl/D60; Sigma-Aldrich) containing protease
and phosphatase cocktail inhibitors. The lysates were centrifuged
at 14,000 × g for 30 min at 4°C. The total protein concentration of
each sample was measured using a MicroBCA Protein Assay Reagent kit
(Pierce Biotechnology, Inc.). Lysates (15 µl) from each line
in the SDS sample buffer were then separated using 10% SDS-PAGE
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) and electroblotted
onto a polyvinylidene fluoride membrane (EMD Millipore, Billerica,
MA, USA), which was then blocked with 5% fat-free milk in
phosphate-buffered saline with 0.1% Tween 20 (Sigma-Aldrich) for 1
h at room temperature. The membrane was then incubated with
anti-Rac1, anti-phosphorylated (p-) ERK and anti-p-p38 overnight at
4°C, followed by incubation with horseradish peroxidase-conjugated
secondary antibodies (1:2,000; Beijing Zhongshan Jinqiao
Biotechnology Co., Ltd., Beijing, China) for 2 h at room
temperature. Specific bands of target proteins were stained with
chemiluminescence reagent (Pierce Biotechnology, Inc.).
Densitometric scanning of the exposed X-ray film (Kodak, Rochester,
NY, USA) was used for semi-quantitative measurement of the protein
bands. Target signals were normalized to those of GAPDH, and
analyzed semi-quantitatively using a Quantity One analysis system
(version 2.0; IBM Corporation, Armock, NY, USA).
Statistical analysis
All statistical analyses were performed using SPSS
13.0 for Windows (SPSS, Inc., Chicago, IL, USA). All data are
presented as the mean ± standard deviation. Comparisons between
groups were performed using one-way analysis of variance and the
Student-Newman-Keuls method. P<0.05 was considered to indicate a
statistically significant difference.
Results
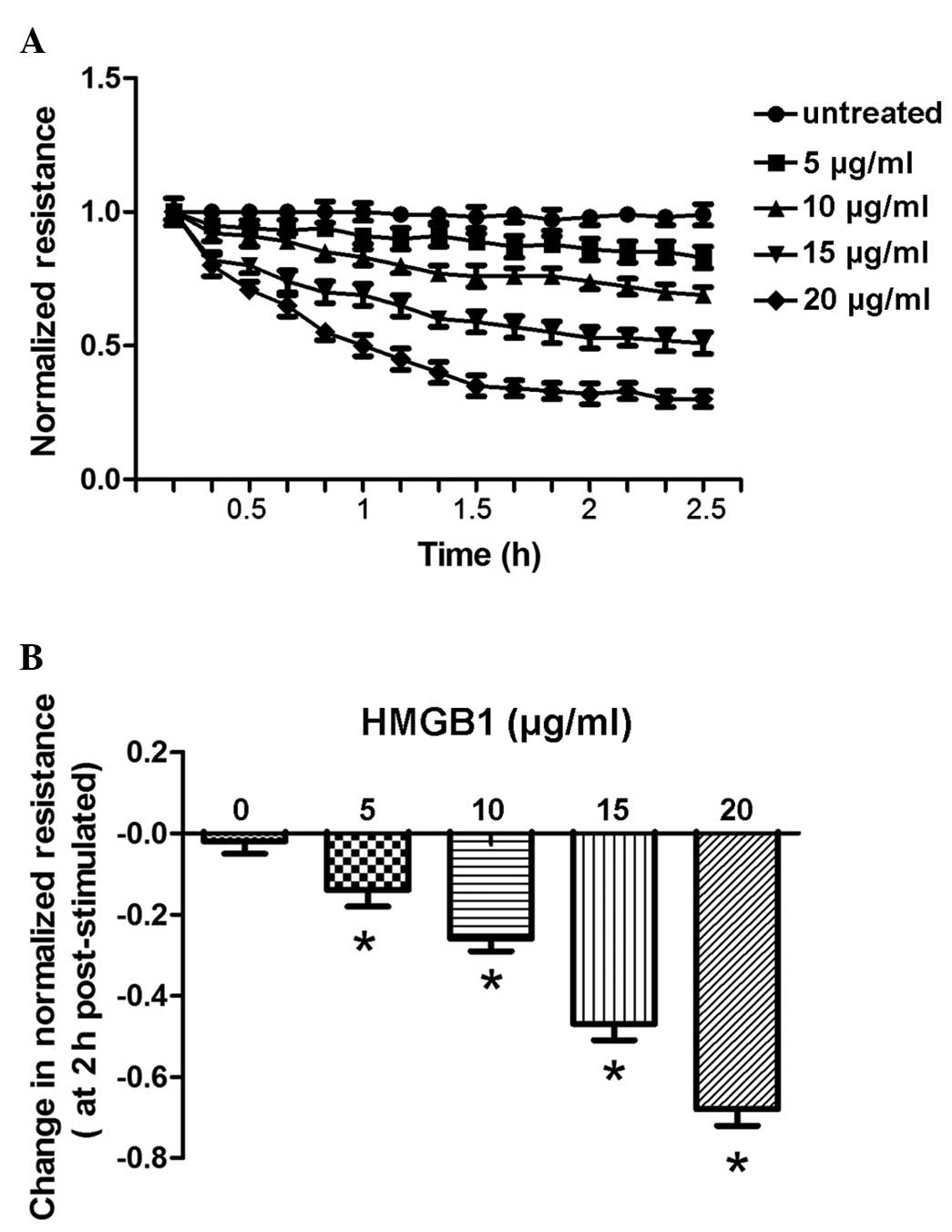
HMGB1 induces dose- and time-dependent
decreases in TER
The present study measured the continuous TER of
cultured PMVECs grown on gold microelectrodes. Challenge with
recombinant HMGB1 produced dose-dependent (5–20 µg/ml) and
time-dependent (0–2.5 h) decreases in TER measurements, indicating
increased EC barrier dysfunction (Fig.
1A), compared with the control buffer, containing 20 mM HEPES
(pH 7.8), 150 mM NaCl, 0.2 mM EDTA, 0.1% Triton X-100, 2 mg/ml
leupeptin and 0.1 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride. The
results also demonstrated that the change in normalized resistance
2 h following stimulation was dose-dependent (5–20 µg/ml;
Fig. 1B).
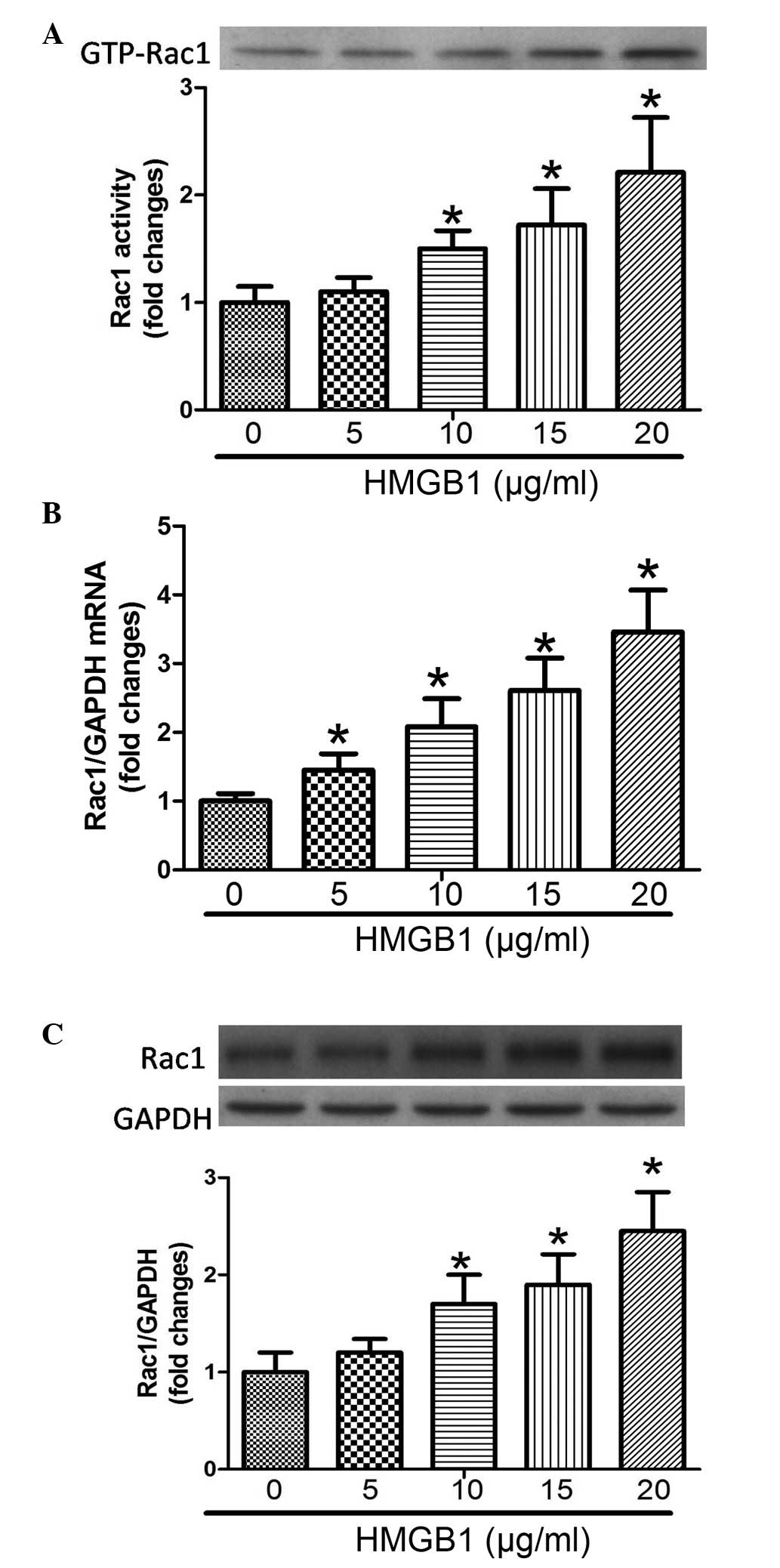
HMGB1 induces a dose-dependent increase
in the expression and activity of Rac1
To the best of our knowledge, there have been no
reports regarding the association between HMGB1 and Rac1,
therefore, the present study aimed to determine whether HMGB1
regulated the expression and activity of Rac1. As shown in Fig. 2, the activity, mRNA expression and
protein expression levels of Rac1 were significantly increased in a
dose-dependent (5–20 µg/ml) manner following challenge of
the PMVECs with HMGB1.
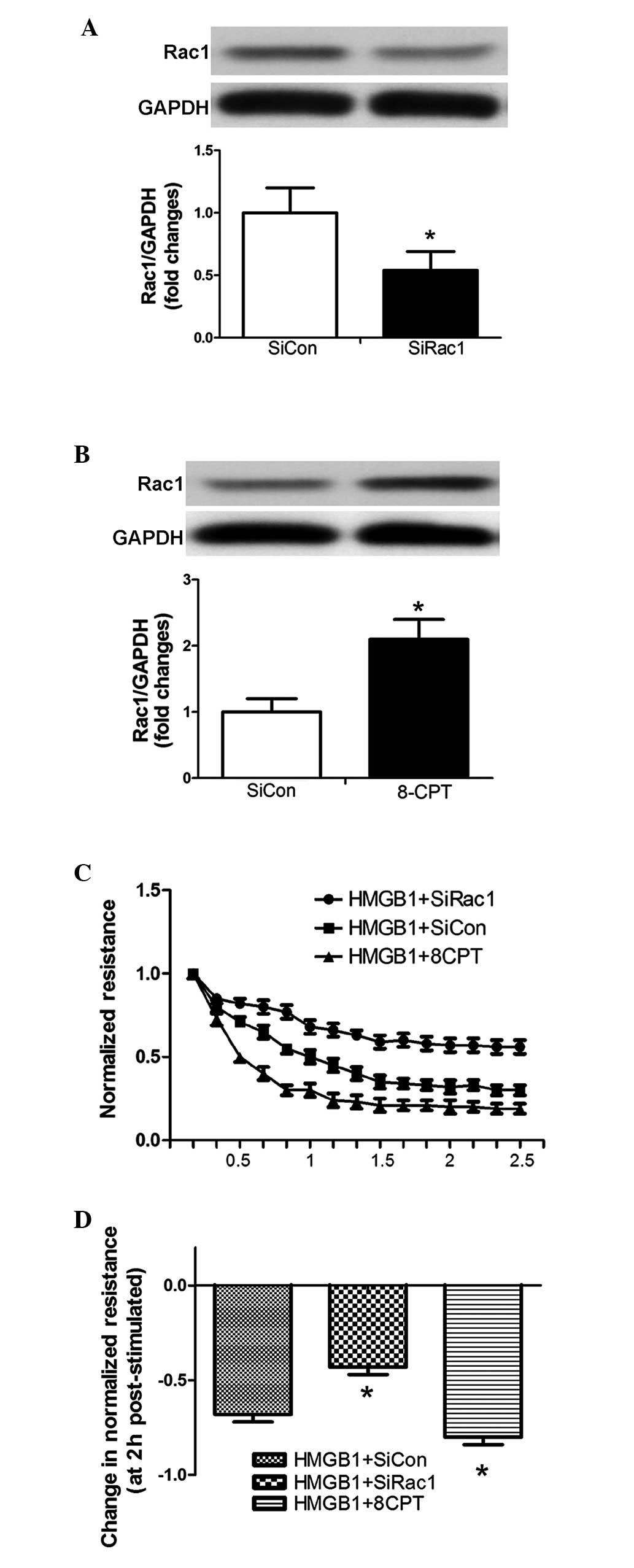
Rac1 is an important mediator of the
HMGB1-induced TER decrease in PMVECs
The siRNA targeting Rac1 and the Rac1 agonist
(8-CPT) were used to inhibit and induce the expression of Rac1 in
PMVECs, respectively (Fig. 3A and
B). The silenced and stimulated cells were subsequently plated
on gold microelectrodes, allowing assay of TER following HMGB1
challenge (20 µg/ml). Downregulation of the expression of
Rac1 significantly attenuated the HMGB1-induced decline in TER,
whereas the overexpression of Rac1 significantly enhanced the
HMGB1-induced decline in TER (Fig. 3C
and D).
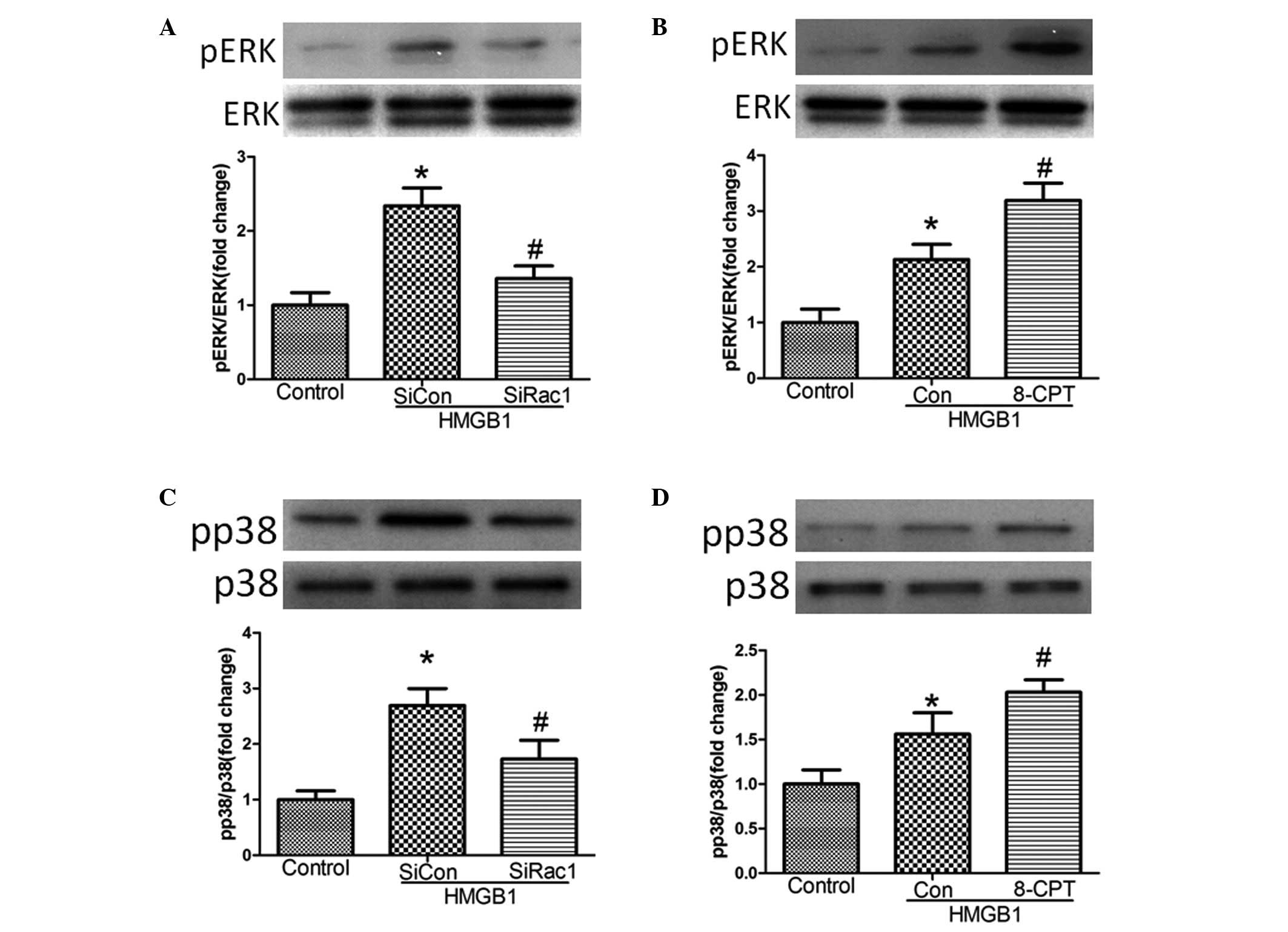
Rac1 mediates MAPK signaling following
HMGB1 challenge
A previous study demonstrated that the MAPK
signaling pathway mediated HMGB1-induced EC barrier disruption
(15). The present investigation
aimed to determine whether Rac1 mediated MAPK signaling following
HMGB1 challenge (20 µg/ml). Treatment of the cells with
siRac1 or 8-CPT significantly inhibited and enhanced the expression
of pERK, respectively, and increased the expression levels of p-p38
following HMGB1 challenge, compared with the control (Fig. 4).
Discussion
The molecular mechanisms underlying the contribution
of HMGB1 to ARDS remain to be fully elucidated. The present study
further demonstrated several molecular events, which are involved
in HMGB1-induced hyperpermeability. The results of the present
study indicated that HMGB1 induced dose- and time-dependent
decreases in TER, and these results are concordant with those of a
previous study (14). Notably,
HMGB1 induced a dose-dependent increase in the activity and
expression levels of Rac1. In addition, following the use of siRNA
and an agonist of Rac1, the results of the present study
demonstrated that Rac1 was a novel factor mediating the
HMGB1-induced decrease in TER via ERK and p38 MAPK activation.
Widespread injury to the lung and systemic
endothelium, resulting in high-permeability pulmonary edema, is a
typical characteristic of ARDS, which was first recognized in
ultra-structural investigations almost 30 years ago (1). Disruption of the semi-permeable
barrier, which is formed by the pulmonary endothelium, results in a
marked increase in the levels of fluid and protein leaving the
vascular space through the alveolar epithelium into the
interstitium and airspaces (1).
The integrity of the pulmonary endothelium barrier is regulated by
competing EC contractile forces and adhesive cell-cell tethering
forces, which are closely associated with the endothelial actin
cytoskeleton. Disruption of actin increases permeability and
damages barrier integrity (2).
Increasing evidence has suggested that HMGB1, a late-acting
cytokine mediating endotoxin-associated lethality, causes
reorganization of the actin cytoskeleton and disruption of an
important junctional protein, vascular endothelial-cadherin, and
induces paracellular gap formation in human pulmonary artery
endothelial cells (15). In the
present study, HMGB1 induced a marked decrease in TER, reflecting
increased barrier dysfunction in PMVECs, and these results are
similar to those of a previous study (15).
There is an increasing body of evidence suggesting
that Rac1, Cdc42 and Rap1 contribute to the maintenance and
stabilization of microvascular endothelial barrier functions,
whereas RhoA primarily acts adversely to impair barrier integrity
(9,17). In previous years, there has been
substantial attention on the role of Rac in the assembly of
inter-endothelial junctions, and its increase in activity during
junction formation (3). In the
absence of vasoactive stimuli, Rac1 deficiency increases
endothelial permeability and cannot form lamellipodial structures,
focal adhesions or cell-cell contacts due to adherens and tight
junction dysfunction, indicating that Rac is important for
maintaining cell-cell junctions (10,18).
Furthermore, a previous study demonstrated that treatment with
NSC-23766, which disturbs the Rac1-specific GEFs T-cell lymphoma
invasion and metastasis 1 and Trio, also decreased TER and promoted
intercellular gap formation (19).
Our previous study also demonstrated that TNF-α-induced PMVECs
barrier breakdown was, at least in part, mediated by Rac1
inactivation (20). In addition,
as the majority of findings on the microvascular endothelium are in
accordance with existing reports that macrovascular endothelial
cells are negatively affected by constitutively inactive Rac1
mutants (9,18), it is well-established that Rac1
appears to improve endothelial barrier properties under resting
conditions. However, several investigations have reported different
results. In vitro, unlike thrombin, VEGF has been observed
to induce rapid activation of Rac1 and increase the production of
ROS, resulting in endothelial barrier dysfunction (8). The activation of Rac1 in the
macrovascular endothelium may induce barrier destabilization,
possibly via the generation of ROS (9–11).
Therefore, the protective or deleterious role of Rac1 activation in
microvascular endothelial barrier function is dependent on cell
type and various stimuli. In the present study, the results
demonstrated that the activity and expression levels of Rac1 in
PMVECs were elevated when stimulated by HMGB1, and Rac1 acted as a
promoter of HMGB1-induced barrier dysfunction and endothelial
hyperpermeability. These findings indicate a preliminary role for
Rac1 in HMGB1-induced barrier dysfunction in PMVECs, and requires
further investigation in the future.
The signaling pathways involved in HMGB1-induced
barrier dysfunction have been investigated in previous studies.
Wolfson et al (15)
demonstrated that the receptor for advanced glycation end products
(RAGE) was the primary receptor signaling HMGB1-induced TER
decrease and paracellular gap formation. Of note, ECs treated with
AGE-bovine serum albumin show increased permeability and actin
cytoskeleton rearrangement, and these effects were alleviated
following treatment with anti-RAGE antibody, specific MAPK
inhibitors or dominant negative forms of ERK and p38 MAPK,
indicating that the MAPK signaling pathway may be involved in
HMGB1/RAGE-induced barrier dysfunction (21). Wolfson et al (15) also reported that pre-treatment with
SB203580 resulted in the attenuation of HMGB1-induced TER
disruption, and p38 MAPK was the direct downstream target of
HMGB1/RAGE. In the present study, the results further demonstrated
that HMGB1 stimulated the expression of p-ERK and p-p38 in PMVECs.
A previous study suggested that Rac induces MAPK signaling under
several conditions. Lin et al (22) reported that treatment of human lung
fibroblasts with CXCL12 caused the activation of Rac1, Rho and ERK,
and the CXCL12-induced increase in ERK phosphorylation was
inhibited by RacN17, a dominant negative mutant of Rac1. Using
siRNA and an agonist of Rac1, the present study also demonstrated
that Rac1 promoted ERK/p38 MAPK signaling in the PMVECs following
challenge with HMGB1. Therefore, HMGB1 induced hyperpermeability in
PMVECs via the Rac1/MAPK signaling pathway.
In conclusion, the results of the present study
confirmed the effects of HMGB1 on endothelial barrier function via
measurement of TER, a reflection of loss of barrier integrity. The
underlying mechanisms may, at least in part, be through
Rac1-mediated MAPK signal pathways being involved in HMGB1-induced
hyperpermeability. Future investigations may further elucidate the
precise mechanisms underlying HMGB1-induced ARDS, and thereby
provide novel therapeutic approaches in the treatment of ARDS.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81070232 and
81270372) and the Natural Science Foundation of Anhui Province
(grant no. 1408085MH170).
References
|
1
|
Ware LB: Pathophysiology of acute lung
injury and the acute respiratory distress syndrome. Semin Respir
Crit Care Med. 27:337–349. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dudek SM and Garcia JG: Cytoskeletal
regulation of pulmonary vascular permeability. J Appl Physiol
(1985). 91:1487–1500. 2001.
|
|
3
|
Beckers CM, van Hinsbergh VW and van Nieuw
Amerongen GP: Driving Rho GTPase activity in endothelial cells
regulates barrier Integrity. Thromb Haemost. 103:40–55. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Waschke J, Baumgartner W, Adamson RH, Zeng
M, Aktories K, Barth H, Wilde C, Curry FE and Drenckhahn D:
Requirement of Rac activity for maintenance of capillary
endothelial barrier properties. Am J Physiol Heart Circ Physiol.
286:H394–H401. 2004. View Article : Google Scholar
|
|
5
|
Adamson RH, Ly JC, Sarai RK, Lenz JF,
Altangerel A, Drenckhahn D and Curry FE: Epac/Rap1 pathway
regulates microvascular hyperpermeability induced by PAF in rat
mesentery. Am J Physiol Heart Circ Physiol. 294:H1188–H1196. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Birukova AA, Fu P, Xing J and Birukov KG:
Rap1 mediates protective effects of iloprost against
ventilator-induced lung injury. J Appl Physiol (1985).
107:1900–1910. 2009. View Article : Google Scholar
|
|
7
|
Spindler V, Schlegel N and Waschke J: Role
of GTPases in control of microvascular permeability. Cardiovasc
Res. 87:243–253. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang Y, Zang QS, Liu Z, Wu Q, Maass D,
Dulan G, Shaul PW, Melito L, Frantz DE, Kilgore JA, et al:
Regulation of VEGF-induced endothelial cell migration by
mitochondrial reactive oxygen species. Am J Physiol Cell Physiol.
301:C695–C704. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wojciak-Stothard B, Tsang LY and Haworth
SG: Rac and Rho play opposing roles in the regulation of
hypoxia/reoxygenation-induced permeability changes in pulmonary
artery endothelial cells. Am J Physiol Lung Cell Mol Physiol.
288:L749–L760. 2005. View Article : Google Scholar
|
|
10
|
van Wetering S, van Buul JD, Quik S, Mul
FP, Anthony EC, ten Klooster JP, Collard JG and Hordijk PL:
Reactive oxygen species mediate Rac-induced loss of cell-cell
adhesion in primary human endothelial cells. J Cell Sci.
115:1837–1846. 2002.PubMed/NCBI
|
|
11
|
Chen W, Pendyala S, Natarajan V, Garcia JG
and Jacobson JR: Endothelial cell barrier protection by
simvastatin: GTPase regulation and NADPH oxidase inhibition. Am J
Physiol Lung Cell Mol Physiol. 295:L575–L583. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang H, Bloom O, Zhang M, Vishnubhakat JM,
Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, et
al: HMG-1 as a late mediator of endotoxin lethality in mice.
Science. 285:248–251. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim DC, Lee W and Bae JS: Vascular
anti-inflammatory effects of curcumin on HMGB1-mediated responses
in vitro. Inflamm Res. 60:1161–1168. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gao M, Hu Z, Zheng Y, Zeng Y, Shen X,
Zhong D and He F: Peroxisome proliferator-activated receptor γ
agonist troglitazone inhibits high mobility group box 1 expression
in endothelial cells via suppressing transcriptional activity of
nuclear factor κB and activator protein 1. Shock. 36:228–234. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wolfson RK, Chiang ET and Garcia JG: HMGB1
induces human lung endothelial cell cytoskeletal rearrangement and
barrier disruption. Microvasc Res. 81:189–197. 2011. View Article : Google Scholar
|
|
16
|
Kim JY, Park JS, Strassheim D, Douglas I,
Diaz del Valle F, Asehnoune K, Mitra S, Kwak SH, Yamada S, Maruyama
I, et al: HMGB1 contributes to the development of acute lung injury
after hemorrhage. Am J Physiol Lung Cell Mol Physiol.
288:L958–L965. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wojciak-Stothard B and Ridley AJ: Rho
GTPases and the regulation of endothelial permeability. Vascul
Pharmacol. 39:187–199. 2002. View Article : Google Scholar
|
|
18
|
Wójciak-Stothard B, Potempa S, Eichholtz T
and Ridley AJ: Rho and Rac but not Cdc42 regulate endothelial cell
permeability. J Cell Sci. 114:1343–1355. 2001.PubMed/NCBI
|
|
19
|
Baumer Y, Spindler V, Werthmann RC,
Bünemann M and Waschke J: Role of Rac 1 and cAMP in endothelial
barrier stabilization and thrombin-induced barrier breakdown. J
Cell Physiol. 220:716–726. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shao M, Yue Y, Sun GY, You QH, Wang N and
Zhang D: Caveolin-1 regulates Rac1 activation and rat pulmonary
micro-vascular endothelial hyperpermeability induced by TNF-α. PLoS
One. 8:e55212013. View Article : Google Scholar
|
|
21
|
Guo XH, Huang QB, Chen B, Wang SY, Li Q,
Zhu YJ, Hou FF, Fu N, Brunk UT and Zhao M: Advanced glycation end
products induce actin rearrangement and subsequent
hyperpermeability of endothelial cells. APMIS. 114:874–883. 2006.
View Article : Google Scholar
|
|
22
|
Lin CH, Shih CH, Tseng CC, Yu CC, Tsai YJ,
Bien MY and Chen BC: CXCL12 induces connective tissue growth factor
expression in human lung fibroblasts through the Rac1/ERK, JNK, and
AP-1 pathways. PLoS One. 9:e1047462014. View Article : Google Scholar : PubMed/NCBI
|