Introduction
Inflammation is induced in response to infectious
diseases, and protects the body from invasion. Macrophages have a
key role in the course of inflammation, which can be activated by
various endogenous and/or exogenous factors. Once activated,
macrophages may release a large amount of inflammatory cytokines
into the extracellular matrix, which further aggravates the
inflammatory response. It has previously been reported that the
expression of numerous genes is induced or suppressed in
macrophages, in response to the activating agent lipopolysaccharide
(LPS) (1). LPS is an outer
membrane glycolipid of gram-negative bacteria, which is recognized
by Toll-like receptor 4 (TLR4) thereby initiating an inflammatory
response (2,3).
TLR4 signaling triggers the activation of various
transcription factors, including nuclear factor (NF)-κB and
mitogen-activated protein kinases, such as extracellular
signal-regulated kinases (4).
NF-κB has a key role in various biological processes, including
immunity, inflammation, wound healing, proliferation and apoptosis
(5). Numerous inflammatory
diseases, including rheumatoid arthritis, multiple sclerosis and
inflammatory bowel disease, are associated with the activation of
NF-κB (6,7).
Acetylation of histones is a type of
post-translational modification, which is regulated by the opposing
activities of histone deacetylases (HDACs) and histone
acetyltransferases (HATs). Histone acetylation is regulated by
HATs, which activate gene transcription by relaxing the structure
of chromatin, whereas deacetylation is controlled by HDACs, which
induce the condensation or inactivation of chromatin, leading to
gene suppression (8,9). In addition to histones, proteins
without histones may also undergo acetylation or deacetylation by
HATs and HDACs (10,11).
Histone deacetylase inhibitors (HDACi) are currently
used in the routine clinical treatment of cancer (12,13).
Alongside the antitumor activity of HDACi, increased attention has
been paid to their anti-inflammatory effects. Theoretically, HDACs
are associated with gene repression via histone inactivation,
whereas HDACi should promote gene transcription and enhance
inflammatory responses. However, it has previously been reported
that HDACi, such as valproic acid, suberoylanilide hydroxamic acid
(SAHA) and Trichostatin A (TSA), are able to suppress the
expression of proinflammatory cytokines and increase the survival
rate of mice following septic shock (14–16).
Therefore, the anti-inflammatory effects of HDACi, and the
underlying mechanisms require further clarification.
TSA is a hydroxamic acid-based compound, which was
originally developed as an antifungal agent (17). TSA selectively inhibits class I and
II mammalian HDACs, but not class III HDACs (18). The U-937 monocyte-like cell line is
widely used in research regarding inflammation, and it can be
differentiated by phorbol myristate acetate (PMA) and activated by
LPS (19). The present study aimed
to analyze the inhibitory effects and potential mechanism of the
HDACi TSA, on the release of inflammatory factors in macrophages
differentiated from U-937 cells.
Materials and methods
Cell culture
U-937 cells (purchased from American Type Culture
Collection, Manassas, VA, USA and preserved in our laboratory) were
cultured and maintained at a cell density between 5×105
and 1×106 cells/ml in RPMI-1640 media (Gibco; Thermo
Fisher Scientific, Inc., Villebon-sur-Yvette, France) supplemented
with 10% fetal bovine serum (Biowest, Nuaillé, France), 100 U/ml
penicillin (Genom Co., Ltd., Hangzhou, China), and 100 μg/ml
streptomycin (Genom Co., Ltd.). The cells were incubated at 37°C in
a humidified atmosphere containing 95% air and 5% CO2.
For macrophage-like cell differentiation, the serum-starved U-937
cells were plated in 6-well plates (Corning, Inc., Corning, NY,
USA) at a density of 5×105 cells/ml and treated with 60
ng/ml PMA (Sigma-Aldrich, St. Louis, MO, USA) for 24 h. The
non-adherent cells were removed by washing twice with
phosphate-buffered saline (PBS).
Experimental design and cell
treatment
The macrophages were divided into three groups: The
control group, the LPS-treated group and the TSA-treated group. For
the TSA-treated group, TSA (Sigma-Aldrich) was added to the medium
2 h prior to treatment with LPS (1 μg/ml; Sigma-Aldrich).
The final concentrations of TSA were 50, 100, 500 and 1,000 nM [TSA
was dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich), and the
final DMSO concentration was <0.4%]. For the LPS-treated group,
an equal dosage of DMSO was added to the medium 2 h prior to
treatment with LPS (1 μg/ml). For the control group, the
same doses of DMSO and normal saline were added to the medium at
the same time point. The super-natants and cells were harvested 24
h after the addition of LPS.
Cell viability test
Cell viability was assessed using the Cell Counting
kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto,
Japan). The cells (2×105 cells/well) were plated in
96-well plates for the viability assay. Experimental procedures
were performed according to the manufacturer's protocol.
Fluorescence-activated cell sorting
(FACS)
The apoptotic rate of the macrophages was measured
by flow cytometry (Cell Lab Quanta SC; Beckman Coulter, Inc., Brea,
CA, USA) using an Annexin V-Fluorescein Isothiocyanate (FITC)
Apoptosis Detection kit (Lianke Biotech Co., Ltd., Hangzhou,
China). Briefly, non-adherent and adherent cells were collected.
The cells were suspended in 500 μl binding buffer and
stained with Annexin V-FITC and propidium iodide (PI) at room
temperature for 5 min in the dark. After removing the unbound
Annexin V-FITC and PI by centrifugation at 2,000 × g at 4°C for 5
min, the cells were resuspended in excess binding buffer. For each
assay, ≥10,000 cells were analyzed by FACS. Data were expressed as
the percentage of non-viable (Annexin V++PI+)
cells.
Analysis of cell supernatants
The levels of tumor necrosis factor (TNF)-α,
interferon (IFN)-γ, interleukin (IL)-10 and IL-18 in the cell
supernatants were measured using enzyme-linked immunosorbent assay
(ELISA) kits (eBioscience, Inc., San Diego, CA, USA), according to
the manufacturer's protocols.
Reverse transcription-polymerase chain
reaction (RT-PCR) assay
Total RNA was extracted from the isolated
macrophages using TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), according to the
manufacturer's protocol. RNA samples were quantified by measuring
the absorbance (A) at 260 and 280 nm using a spectrophotometer
(UV762; Shanghai Analysis Instrument Co., Ltd., Shanghai, China).
The concentrations of RNA were calculated according to A260.
Aliquots of total RNA (1 μg) from each sample were reverse
transcribed into cDNA, according to the instructions of the First
Strand cDNA Synthesis kit (Takara Bio, Inc., Otsu, Japan).PCR
amplification was performed in a volume of 20 μl containing
2 μl cDNA, 0.25 μmol/l of each primer, 0.25 mmol/l
deoxyribonucleotide triphosphates, and 1 U Takara TaqHS polymerase
(Takara Bio, Inc.) using SYBR Green (Takara Bio, Inc.) as the
fluorescence indicator on a Bio-Rad Cycler system (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). In the PCR amplification
reaction, the thermocycling conditions were as follows: Initial
activation at 95°C for 15 min, followed by 40 cycles of
denaturation at 94°C for 15 sec, annealing for 30 sec at 55°C and
extension for 30 sec at 72°C. The primers used were synthesized by
Sangon Biotech Co., Ltd. (Shanghai, China), and the sequences were
as follows. HDAC1, forward TAA TAA GCA GCA GAC GGA CATCG, reverse
CAT AAT ACT TGC CTT TGC CAGC; HDAC2, forward TGA TGG AGA TGT ATC
AAC CTA GTGC, reverse TTG AAA CAA CCC AGT CTA TCA CCAG; HDAC3,
forward TGG TTA TAC TGT CCG AAA TGT TGC, reverse CTG TCA TAG GTC
AGG AGG TCTGC; cyclooxygenase (COX)-2, forward AAA TCC TTG CTG TTC
CCACC, reverse TTT CTC CAT AGA ATC CTG TCCG; chemokine (C-X-C
motif) ligand 2 (CXCL-2), forward AAA GTG TGA AGG TGA AGT CCCCC,
reverse GTG ATG CTC AAA CAC ATT AGGCG; chemokine (C-C motif) ligand
7 (CCL-7), forward ACA GAA GGA CCA CCA GTA GCCAC, reverse GCT TTG
GAG TTT GGG TTT TCT TGT; and GAPDH, forward CCACATCGCTCAGACACCAT,
reverse CCAGGCGCCCAATACG. The quantification cycle (Cq) value was
analyzed from the amplification plots, and gene expression was
normalized against the Ct of the GAPDH housekeeping gene using the
2−ΔΔCt method (20).
Western blot assay
The U-937 cells were washed twice with cold PBS, and
lysed with lysis buffer (Beyotime Institute of Biotechnology,
Beijing, China) on ice for 1 h. Following centrifugation at 12,000
× g at 4°C for 5 min, the supernatants were collected. The
concentrations of protein were measured with a bicinchoninic acid
protein assay kit (Beyotime Institute of Biotechnology). Briefly,
50 μg protein extracts were separated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred onto a
polyvinylidene fluoride membrane (EMD Millipore, Billerica, MA,
USA). The membranes were blocked with 5% skim milk for 1 h and then
incubated with the following polyclonal primary antibodies at 4°C
overnight: Anti-HDAC1 (cat. no. 2062; 1:1,000; Cell Signaling
Technology, Inc., Danvers, MA, USA), anti-HDAC2 (cat. no. 2540;
1:1,000; Cell Signaling Technology, Inc.), anti-HADC3 (cat. no.
2632; 1:1,000; Cell Signaling Technology, Inc.), anti-H2A (cat. no.
2578; 1:1,000; Cell Signaling Technology, Inc.), anti-AH2A (cat.
no. 2576; 1:1,000; Cell Signaling Technology, Inc.), anti-H2B (cat.
no. 2934; 1:1,000; Cell Signaling Technology, Inc.), anti-AH2B
(cat. no. 2571; 1:1,000; Cell Signaling Technology, Inc.), anti-H3
(cat. no. sc-8654; 1:500; Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA), anti-AH3 (cat. no. 8172; 1:1,000; Cell Signaling
Technology, Inc.), anti-H4 (cat. no. 2935; 1:1,000; Cell Signaling
Technology, Inc.), anti-AH4 (cat. no. 2591; 1:1,000; Cell Signaling
Technology, Inc.), anti-p65 (cat. no. 6956; 1:1,000; Cell Signaling
Technology, Inc.), anti-phosphorylated-p65 (cat. no. 3031; 1:1,000;
Cell Signaling Technology, Inc.), anti-Ac-p65 (cat. no. 3045;
1:1,000; Cell signaling Technology, Inc.), anti-TLR4 (cat. no.
sc-10741; 1:500; Santa Cruz Biotechnology, Inc.) and anti-β-actin
(cat. no. 4967; 1:1,000; Cell Signaling Technology, Inc.). This was
followed by incubation with a fluorescent secondary antibody
(LI-COR Biosciences, Lincoln, NE, USA) at 37°C for 2 h. The blot
was analyzed using the Odyssey Infrared Imaging system (LI-COR
Biosciences). Membranes were also probed for β-actin (Cell
Signaling Technology, Inc.) and histone 3 (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) as additional loading
controls.
Statistical analysis
Statistical analysis was performed using SPSS
version 17.0 software for Windows (SPSS, Inc., Chicago, IL, USA).
Data are presented as the mean ± standard deviation, and
comparisons were made using Student's t-test. P<0.05 was
considered to indicate a statistically significant difference.
Results
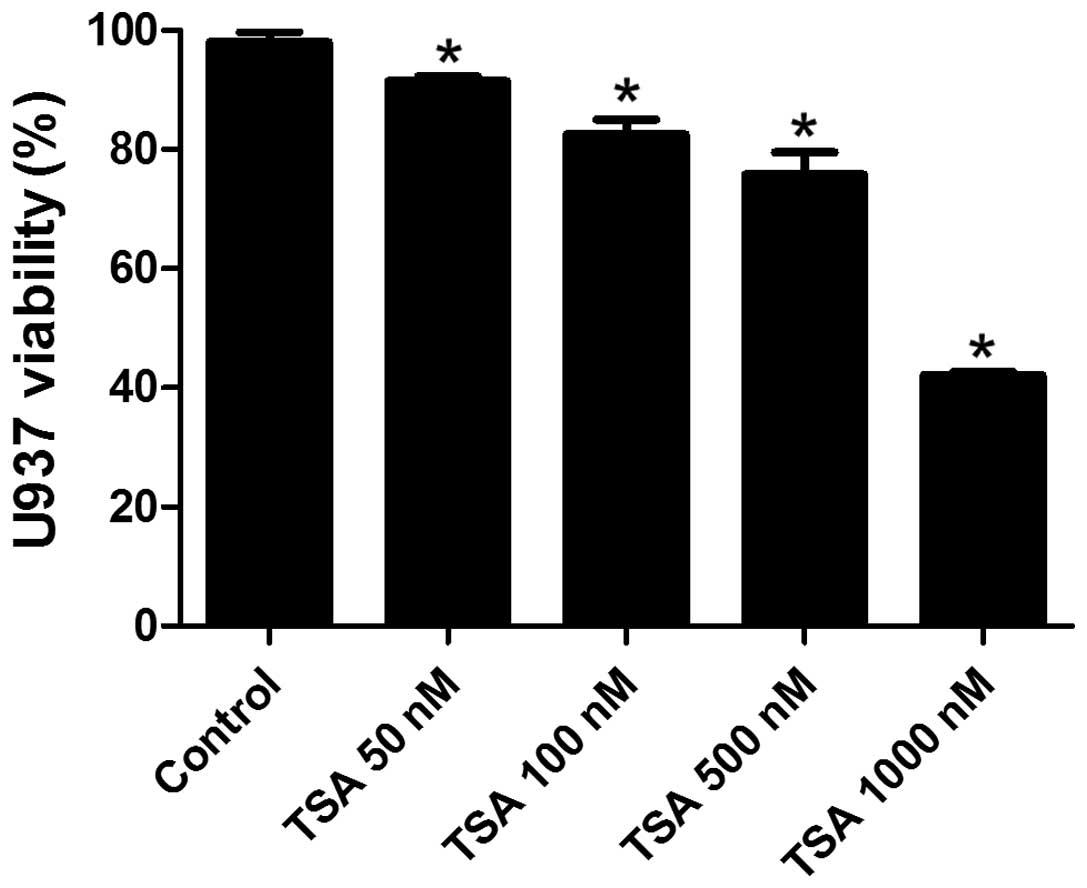
TSA decreases the viability of
PMA-induced macrophages in a dose-dependent manner
In order to determine a suitable treatment dosage of
TSA for the present study, the cytotoxic effects of TSA on the
PMA-induced macrophages were detected by a CCK-8 assay. As shown in
Fig. 1, the viability of
macrophages decreased in a dose-dependent manner when exposed to
various doses of TSA (50, 100, 500 and 1,000 nM). The cell
viability was 93% when treated with 50 nM TSA, whereas the
viability decreased to 42% when the cells were treated with 1,000
nM TSA. Cell viability was >80% when the dose of TSA was below
500 nM; therefore, the present study used 50, 100 and 500 nM TSA to
explore the effects of TSA on macrophages.
TSA decreases the levels of TNF-α, IFN-γ,
IL-10 and IL-18 in macrophages
The expression levels of TNF-α, IFN-γ, IL-10 and
IL-18 were measured in the cell supernatants by ELISA. As shown in
Fig. 2, TNF-α, IFN-γ, IL-10 and
IL-18 expression levels were significantly increased in the
LPS-treated group, as compared with the control group (P<0.05).
The expression levels of TNF-α, IFN-γ, IL-10 and IL-18 were
markedly decreased in the TSA-treated group, as compared with the
LPS-treated group (P<0.05), in a dose-dependent manner.
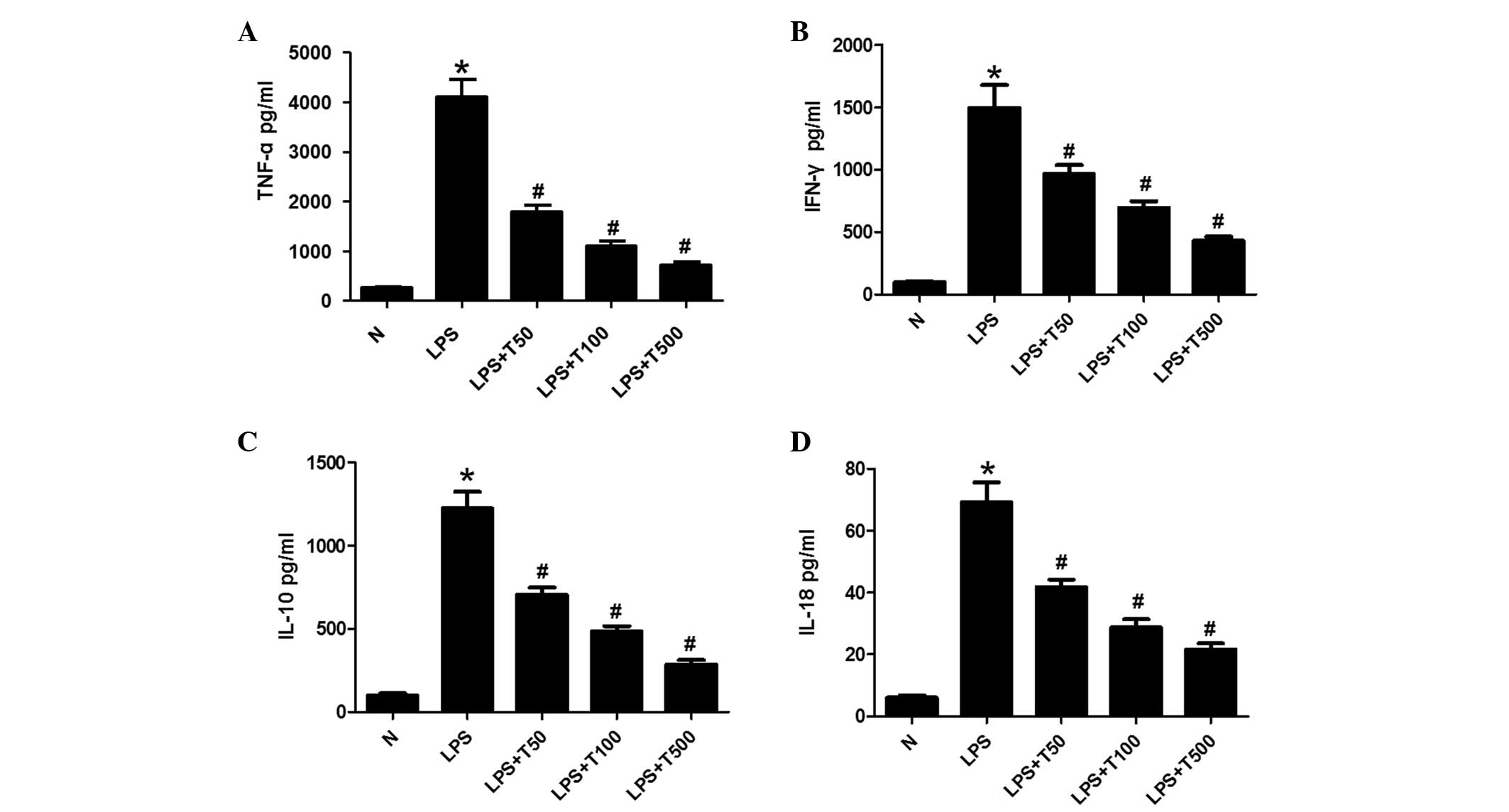 | Figure 2TSA decreased the expression levels of
TNF-α, IFN-γ, IL-10 and IL-18 in macrophages. (A) TNF-α, (B) IFN-γ,
(C) IL-10 and (D) IL-18 expression levels were significantly
increased in the supernatants of the LPS-treated cells, as compared
with the control group. The expression levels of TNF-α, IFN-γ,
IL-10 and IL-18 were markedly decreased in the supernatants of the
TSA-treated cells, as compared with the LPS-treated group, in a
dose-dependent manner. Data are presented as the mean ± standard
deviation. *P<0.05, vs. the control group;
#P<0.05, vs. the LPS-treated group. N, control group;
LPS, lipopolysaccharide; TSA, Trichostatin A; TNF, tumor necrosis
factor; IFN, interferon; IL, interleukin. |
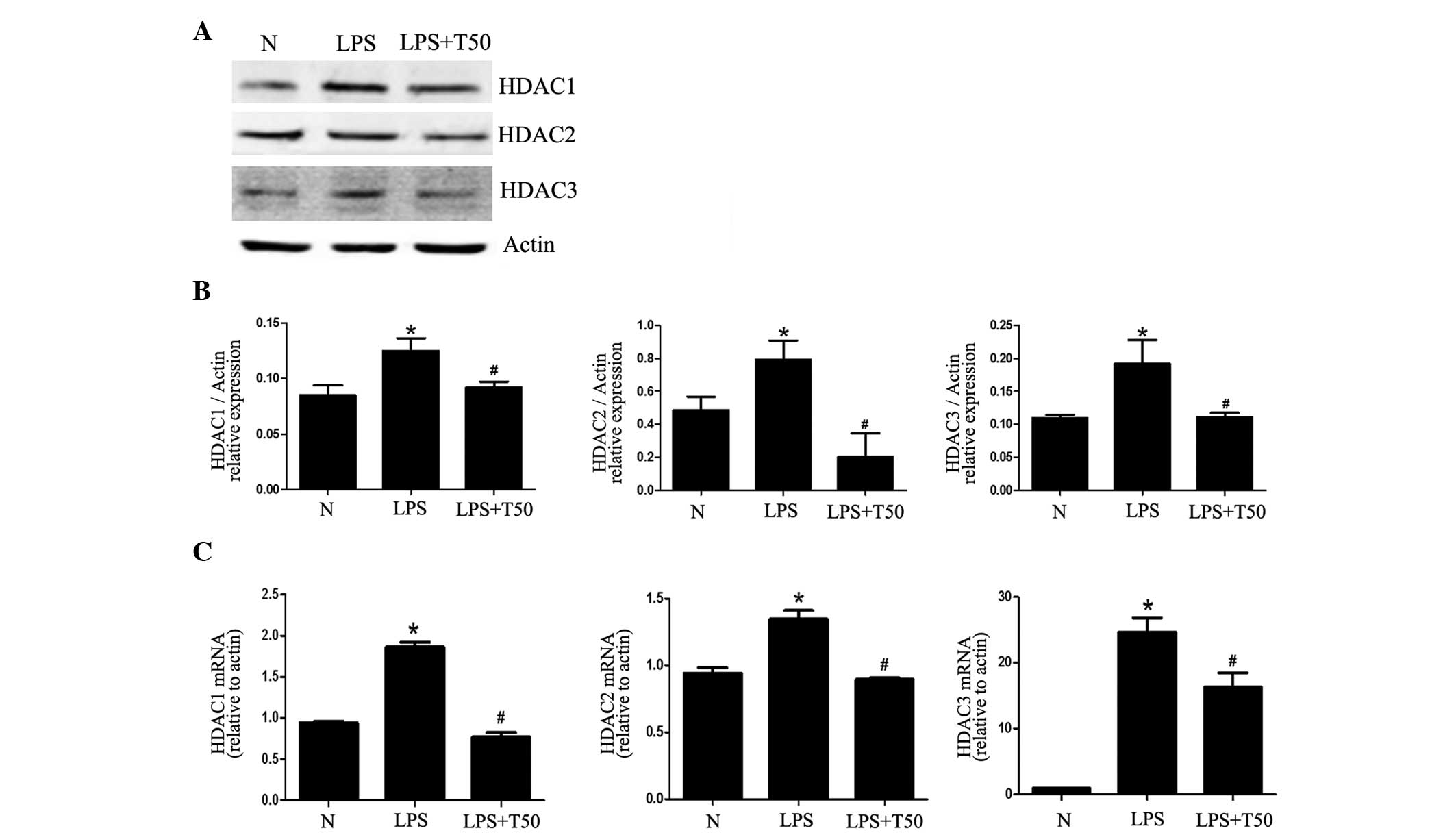
TSA suppresses the expression of class I
HDACs in macrophages
To detect changes in the expression of the
substrates of TSA, the mRNA and protein expression levels of class
I HDACs (HDAC1, HDAC2 and HDAC3) were detected by RT-PCR and
western blotting, respectively. As shown in Fig. 3, the expression levels of HDAC1,
HDAC2 and HDAC3 were markedly increased in the LPS-treated group,
as compared with the normal group; however, the expression levels
were all decreased in the TSA-treated group. These results suggest
that the expression of class I HDACs may be enhanced in the process
of inflammation, and may be decreased by TSA alongside the
inhibition of inflammation.
Low dose treatment with TSA (50 nM) does
not induce apoptosis of macrophages
As shown in Fig. 4,
as compared with the PMA-induced macrophages in normal conditions
(Fig. 4A), a low dose of TSA (50
nM) had little effect on the apoptosis of macrophages (Fig. 4B). However, there was obvious
apoptosis in the macrophages that were treated with higher doses of
TSA (100 or 500 nM, respectively) (Fig. 4C and D). When LPS was added
(Fig. 4E–H), as compared with the
normal condition, the survival rate of the macrophages was
significantly decreased (79.1 vs. 92.4%) (Fig. 4E). However, when treated with 50 nM
TSA, the survival rate of the macrophages was increased to 88.2%
(Fig. 4F). In addition, when
treated with higher doses of TSA (100 or 500 nM respectively), the
survival rate of the macrophages decreased to 67.3 and 52.4%,
respectively (Fig. 4G and H).
These results suggest that the inhibitory effects of low dose TSA
(50 nM) on the release of inflammatory cytokines are not due to
TSA-induced apoptosis of macrophages.
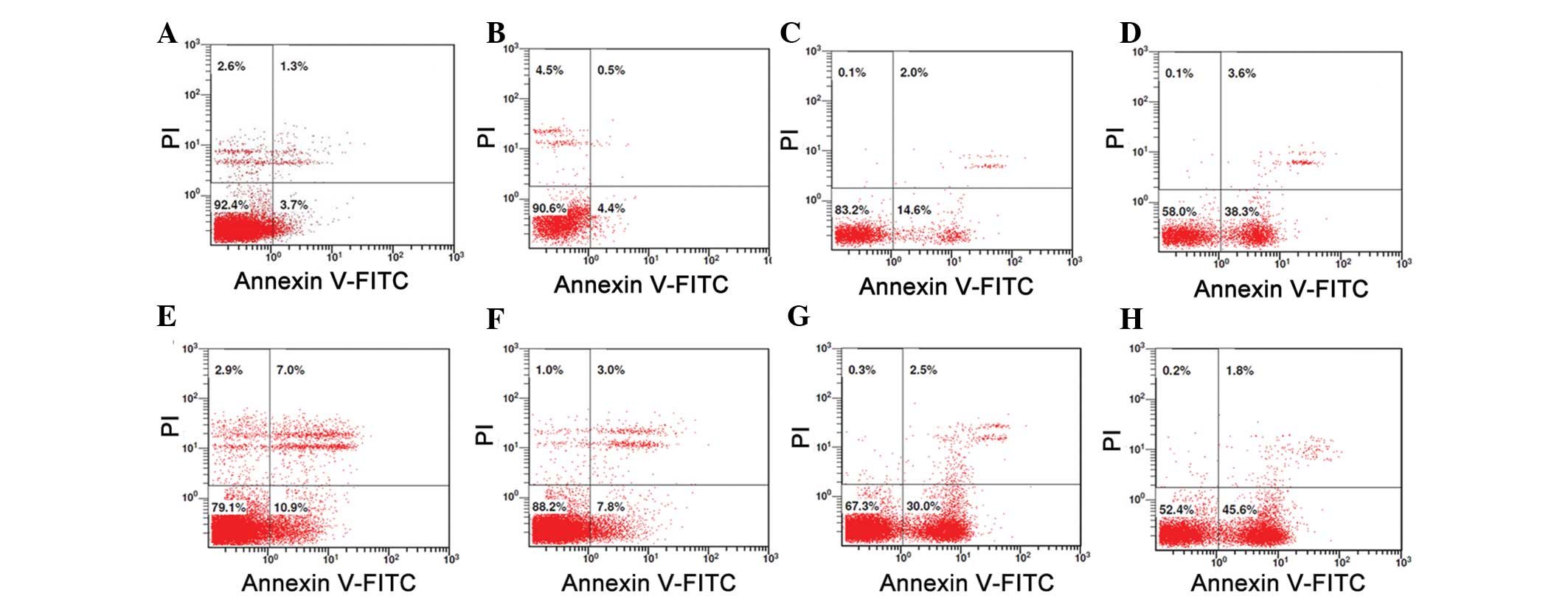 | Figure 4Low dose of TSA (50 nM) did not induce
apoptosis of the macrophages. (A) Compared with the PMA-induced
macrophages in normal conditions (control), (B) the low dose of TSA
(50 nM) had little effect on apoptosis. However, there was marked
apoptosis in the macrophages that were treated with (C) 100 nM or
(D) 500 nM of TSA. (E) Compared with the control, the survival rate
of the macrophages was significantly decreased (79.1 vs. 92.4%)
without TSA treatment. (F) When treated with 50 nM TSA, the
survival rate of the macrophages was increased to 88.2%. However,
when treated with (G) 100 nM TSA or (H) 500 nM TSA, the survival
rate of the macrophages was decreased to 67.3 and 52.4%,
respectively. TSA, Trichostatin A; PMA, phorbol myristate acetate;
FITC, fluorescein isothiocyanate; PI, propidium iodide. |
TSA selectively suppresses the expression
of proinflammatory genes in macrophages
The present study detected the mRNA expression
levels of COX-2, CXCL-2 and CCL-7, in order to explore whether
LPS-induced gene expression could be suppressed by a low dose of
TSA (50 nM). As shown in Fig. 5,
the mRNA expression levels of COX-2, CXCL-2 and CCL-7 were
significantly increased in the LPS-treated group, as compared with
the normal group (P<0.05). In addition, COX-2 and CXCL-2 mRNA
expression levels were significantly increased in the TSA-treated
group, whereas CCL-7 mRNA expression levels were decreased, as
compared with the LPS-treated group (P<0.05). These results
suggest that TSA may differentially regulate the expression levels
of specific proinflammatory genes in macrophages.
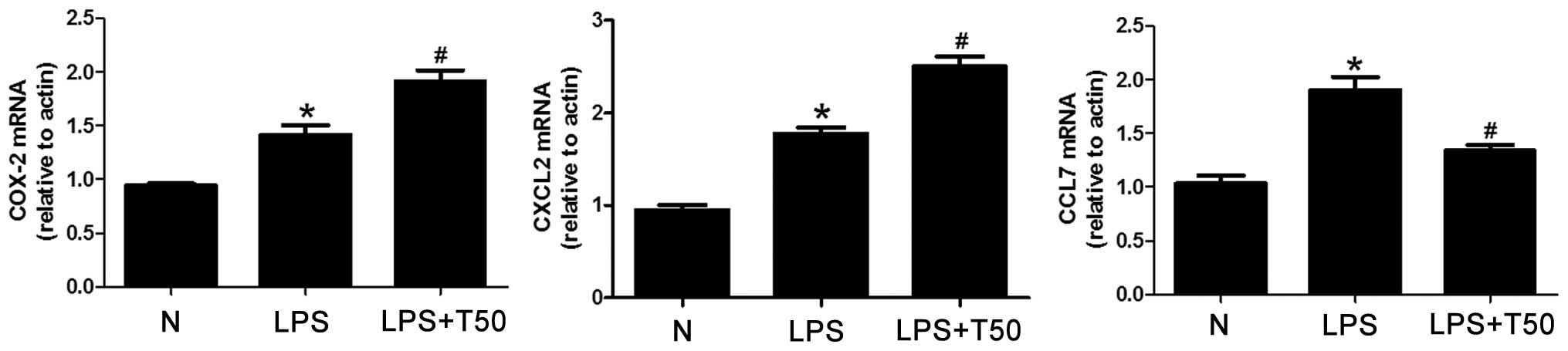 | Figure 5TSA selectively suppressed the
expression levels of proinflammatory genes in macrophages. The mRNA
expression levels of COX-2, CXCL-2 and CCL-7 were significantly
increased in the LPS-treated group, as compared with the normal
group. COX-2 and CXCL-2 mRNA expression levels were significantly
increased in the TSA-treated group, whereas CCL-7 mRNA expression
levels were decreased, as compared with the LPS-treated group. Data
are presented as the mean ± standard deviation.
*P<0.05, vs. the control group;
#P<0.05, vs. the LPS-treated group. N, control group;
LPS, lipopolysaccharide; TSA, Trichostatin A; COX, cyclooxygenase;
CXCL2, chemokine (C-X-C motif) ligand 2; CCL6, chemokine (C-C
motif) ligand 7. |
TSA promotes histone acetylation in
macrophages
To investigate whether the inhibitory effects of TSA
on inflammation were due to the acetylation of histones, the
acetylation levels of histones was determined. As shown in Fig. 6, the acetylation levels of histone
(H)2A, H2B, H3 and H4 were significantly increased in the
LPS-treated group, as compared with the normal group (P<0.05).
Furthermore, the acetylation levels of H2A, H2B, H3 and H4 were all
significantly increased in the TSA-treated group (P<0.05). These
results suggest that the inhibitory effects of TSA on inflammation
may be caused by enhancement of histone acetylation, in order to
selectively suppress the expression of proinflammatory genes.
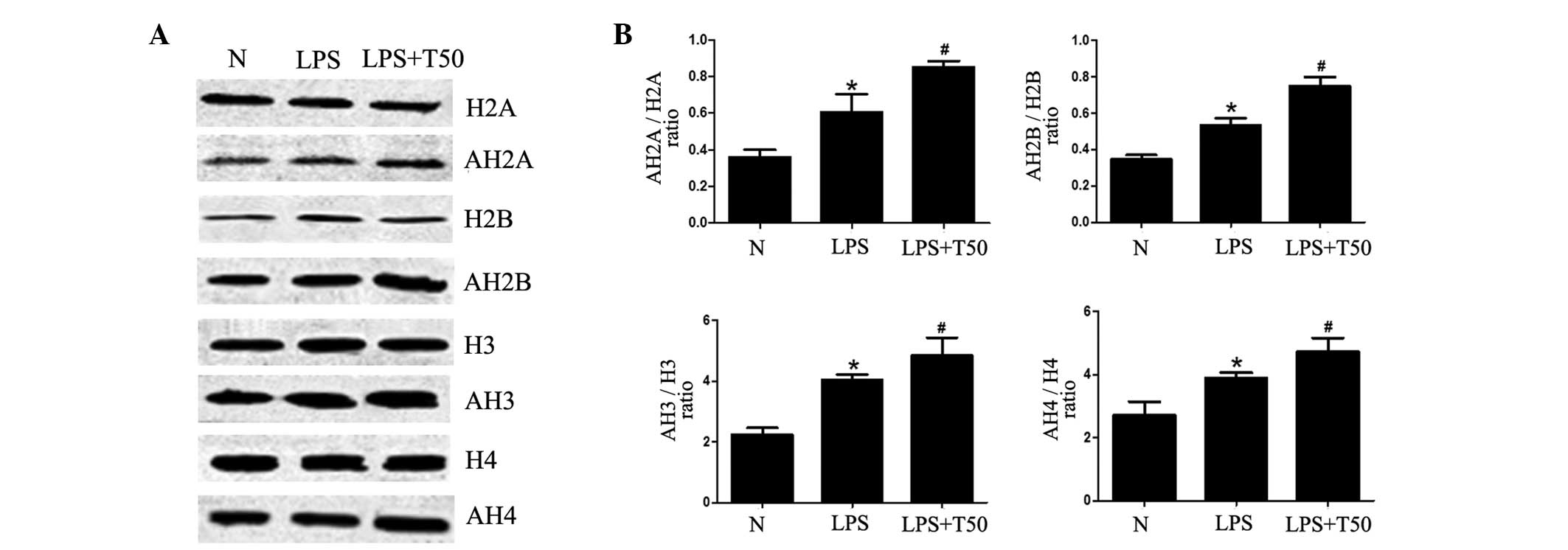 | Figure 6(A) TSA promoted histone acetylation
in macrophages. (B) Acetylation levels of H2A, H2B, H3 and H4 were
all significantly increased in the LPS-treated group, as compared
with the normal group. Furthermore, the acetylation levels of H2A,
H2B, H3 and H4 were all significantly increased in the TSA-treated
group. N, control group; LPS, LPS-treated group; TSA, TSA-treated
group. Data are presented as the mean ± standard deviation.
*P<0.05, vs. the control group;
#P<0.05, vs. the LPS-treated group. N, control group;
LPS, lipopolysaccharide; TSA, Trichostatin A; H, histone; AH,
acetylated histone. |
TSA enhances the acetylation of NF-κB p65
in macrophages
The present study determined whether the inhibitory
effects of TSA on inflammation were via regulation of the
TLR4/NF-κB pathway. As shown in Fig.
7, the protein levels of TLR4 in the LPS-treated group were
significantly higher, as compared with the normal group
(P<0.05), whereas they were decreased in the TSA-treated group.
In addition, there were alterations in the acetylation and
phosphorylation of NF-κB p65. The acetylation of NF-κB p65 was
significantly decreased in the LPS-treated group, as compared with
the normal group, but was significantly increased in the
TSA-treated group (P<0.05). Conversely, the phosphorylation of
NF-κB p65 was increased in the LPS-treated group, but was decreased
in the TSA-treated group (P<0.05). These results suggest that
TSA may affect the TLR4/NF-κB p65 signaling pathway by regulating
the balance of acetylation and phosphorylation of NF-κB p65.
Therefore, the TSA-induced inhibition of inflammation may be due to
regulation of the acetylation of non-histone proteins.
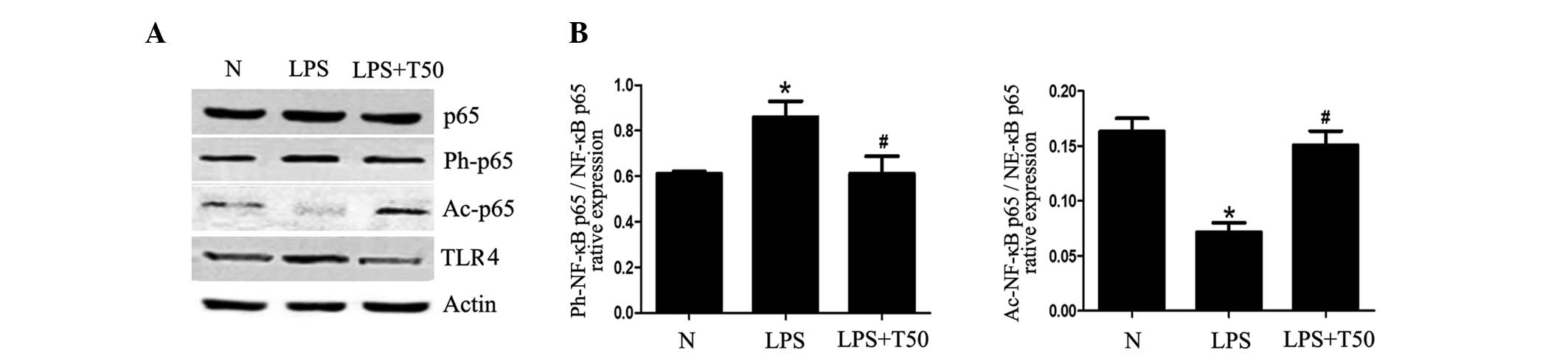 | Figure 7(A) TSA enhanced the acetylation of
NF-κB p65 in macrophages. (B) Expression levels of TLR4 were
significantly higher in the LPS-treated group, as compared with the
normal group, and were decreased in the TSA-treated group. There
were alterations in the acetylation and phosphorylation of NF-κB
p65. The acetylation of NF-κB p65 was significantly decreased in
the LPS-treated group, as compared with the normal group, but was
significantly increased in the TSA-treated group. Conversely, the
phosphorylation of NF-κB p65 was increased in the LPS-treated
group, but was decreased in the TSA-treated group. Data are
presented as the mean ± standard deviation. *P<0.05,
vs. the control group; #P<0.05, vs. the LPS-treated
group. N, control group; LPS, lipopolysaccharide; TSA, Trichostatin
A; NF, nuclear factor; Ph, phosphorylated; Ac, acetylated; TLR4,
Toll-like receptor 4. |
Discussion
Epigenetic mechanisms have been identified as a
major determinant of gene expression and have been implicated in
the regulation of complex physiological and pathological processes.
In addition to methylation, histone acetylation is considered a key
principle of epigenetic regulation. The present study explored the
inhibitory effects of the HDACi TSA, on the release of inflammatory
mediators from macrophages differentiated from U-937 cells.
Since TSA is currently considered an antitumor
agent, the cytotoxicity of TSA was initially detected in the
present study. Cell viability was >80% when treated with <500
nM TSA; therefore, the present study used 50, 100 and 500 nM TSA to
explore the effects of TSA on macrophages. The inhibitory effects
of TSA on the release of inflammatory cytokines were also
investigated. The results of the present study demonstrated that
TSA was able to reduce the cell supernatant expression levels of
TNF-α, IFN-γ, IL-10 and IL-18 in a dose-dependent manner. FACS
assay further demonstrated that the TSA-induced suppression of
inflammatory factors was not due to the apoptosis of macrophages.
These results indicated that the inhibitory effects of TSA on
inflammation were independent of macrophage cell death.
Histone acetylation is controlled by HATs and HDACs.
At present, 18 members of the HDAC family have been identified
(21). The class I (HDAC1, 2, 3
and 8) and class II (HDAC4, 5, 6, 7, 9, 10 and 11) isoforms are
Zn-dependent, whereas the class III HDACs (Sirtuins1, 2 and 7) are
NAD+-dependent.
TSA and SAHA have a hydroxamate structure, and are
able to inhibit Zn-dependent HDAC isoforms (21,22).
In the present study, the expression levels of TSA substrates,
HDAC1, HDAC2 and HDAC3, were detected. The results demonstrated
that the expression levels of HDAC1, HDAC2 and HDAC3 were increased
in the macrophages undergoing LPS-induced inflammation, and were
decreased following TSA treatment. These results suggested that the
expression levels of class I HDACs were enhanced during the process
of inflammation, and could be weakened by TSA alongside the
inhibition of inflammation.
It has previously been reported that the expression
levels of COX-2, CXCL-2 and CCL-7 are increased in LPS-induced
inflammation (23). In the present
study, the expression levels of COX-2, CXCL-2 and CCL-7 were
detected; the mRNA expression levels of COX-2, CXCL-2 and CCL-7
were all increased in the LPS-treated group. However, only the
expression levels of CCL-7 could be reduced by TSA. These results
suggested that TSA may selectively suppress the expression of
proinflammatory genes.
The nucleosome comprises an octamer of four core
histones, an H3/H4 tetramer and two H2A/H2B dimers, which are
surrounded by DNA (146 bp) (24).
This architecture of chromatin is effectively regulated by histone
acetylation. In the present study, histone acetylation was
detected. The results demonstrated that the acetylation of H2A,
H2B, H3 and H4 were all increased following the induction of
inflammation by LPS. Furthermore, when treated with TSA, the
acetylation levels of H2A, H2B, H3 and H4 were further increased.
These results indicated that TSA promoted the acetylation, instead
of inhibiting the acetylation, of histones. Previous studies have
demonstrated that the acetylation of histones is closely associated
with the activation of gene transcription (25–28),
which may promote inflammation by enhancing the expression of
proinflammatory genes (29,30).
However, the results of the present study suggested that if the
levels of histone acetylation were increased following treatment
with the HDACi TSA, the expression of proinflammatory genes could
be selectively suppressed.
In addition to histones, non-histone proteins may
also be regulated by HATs and HDACs. In the present study, the
acetylation levels of NF-κB p65 were measured. The results
demonstrated that the acetylation of NF-κB p65 was decreased in the
LPS-treated group and increased in the TSA-treated group. In
addition to acetylated-NF-κB p65, the levels of TLR4 and
phosphorylated-NF-κB p65 were detected. The results demonstrated
that the expression levels of TLR4 and phosphorylated-NF-κB p65
were decreased following TSA treatment. These results suggested
that TSA may predominantly inhibit the inflammatory response by
regulating the acetylation of non-histone proteins. The specific
mechanism underlying this effect requires further
investigation.
The exact mechanism underlying the anti-inflammatory
effects of TSA, and whether histone or non-histone-mediated effects
are the most important remains unclear. Research regarding the
development of HDAC-selective inhibitors and/or knockout mice may
aid in resolving uncertainties.
In conclusion, the present study demonstrated that
TSA may inhibit inflammation in macrophages. However, whether the
mechanism by which TSA inhibits inflammation is through
significantly enhancing histone acetylation to selectively suppress
the expression of proinflammatory genes, and/or through regulating
non-histone acetylation, requires further research.
Acknowledgments
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no.
81371789).
Abbreviations:
|
HDACi
|
histone deacetylase inhibitors
|
|
TSA
|
trichostatin A
|
|
LPS
|
lipopolysaccharide
|
|
NF-κB
|
nuclear factor κB
|
|
MAPKs
|
mitogen-activated protein kinases
|
|
TLR4
|
toll like receptor 4
|
|
HDACs
|
histone deacetylases
|
|
HATs
|
histone acetyltransferases
|
|
PMA
|
phorbol myristate acetate
|
|
PBS
|
phosphate-buffered saline
|
|
FACS
|
fluorescence-activated cell
sorting
|
|
CXCL-2
|
chemokine (C-X-C motif) ligand 2
|
|
CCL-7
|
chemokine (C-C motif) ligand
|
|
COX-2
|
cyclooxygenase 2
|
References
|
1
|
Wells CA, Ravasi T, Faulkner GJ, Carninci
P, Okazaki Y, Hayashizaki Y, Sweet M, Wainwright BJ and Hume DA:
Genetic control of the innate immune response. BMC Immunol.
4:52003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Beutler B and Rietschel ET: Innate immune
sensing and its roots: The story of endotoxin. Nat Rev Immunol.
3:169–176. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Park BS, Song DH, Kim HM, Choi BS, Lee H
and Lee JO: The structural basis of lipopolysaccharide recognition
by the TLR4-MD-2 complex. Nature. 458:1191–1195. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Régnier CH, Song HY, Gao X, Goeddel DV,
Cao Z and Rothe M: Identification and characterization of an
IkappaB kinase. Cell. 90:373–383. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Karin M and Lin A: NF-kappaB at the
crossroads of life and death. Nat Immunol. 3:221–227. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li Q and Verma IM: NF-kappaB regulation in
the immune system. Nat Rev Immunol. 2:725–734. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huxford T, Huang DB, Malek S and Ghosh G:
The crystal structure of the IkappaBalpha/NF-kappaB complex reveals
mechanisms of NF-kappaB inactivation. Cell. 95:759–770. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Forsberg EC and Bresnick EH: Histone
acetylation beyond promoters: Long-range acetylation patterns in
the chromatin world. Bioessays. 23:820–830. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wade PA: Transcriptional control at
regulatory checkpoints by histone deacetylases: Molecular
connections between cancer and chromatin. Hum Mol Genet.
10:693–698. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hubbert C, Guardiola A, Shao R, Kawaguchi
Y, Ito A, Nixon A, Yoshida M, Wang XF and Yao TP: HDAC6 is a
microtubule-associated deacetylase. Nature. 417:455–458. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Juan LJ, Shia WJ, Chen MH, Yang WM, Seto
E, Lin YS and Wu CW: Histone deacetylases specifically
down-regulate p53-dependent gene activation. J Biol Chem.
275:20436–20443. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Galli M, Salmoiraghi S, Golay J, Gozzini
A, Crippa C, Pescosta N and Rambaldi A: A phase II multiple dose
clinical trial of histone deacetylase inhibitor ITF2357 in patients
with relapsed or progressive multiple myeloma. Ann Hematol.
89:185–190. 2010. View Article : Google Scholar
|
|
13
|
Duvic M, Talpur R, Ni X, Zhang C, Hazarika
P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM and Frankel
SR: Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic
acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood.
109:31–39. 2007. View Article : Google Scholar
|
|
14
|
Cao W, Bao C, Padalko E and Lowenstein CJ:
Acetylation of mitogen-activated protein kinase phosphatase-1
inhibits Toll-like receptor signaling. J Exp Med. 205:1491–1503.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li Y, Liu B, Zhao H, Sailhamer EA,
Fukudome EY, Zhang X, Kheirbek T, Finkelstein RA, Velmahos GC,
deMoya M, et al: Protective effect of suberoylanilide hydroxamic
acid against LPS-induced septic shock in rodents. Shock.
32:517–523. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang L, Wan J, Jiang R, Wang W, Deng H,
Shen Y, Zheng W and Wang Y: Protective effects of trichostatin A on
liver injury in septic mice. Hepatol Res. 39:931–938. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tsuji N, Kobayashi M, Nagashima K,
Wakisaka Y and Koizumi K: A new antifungal antibiotic,
trichostatin. J Antibiot (Tokyo). 29:1–6. 1976. View Article : Google Scholar
|
|
18
|
Okamoto H, Fujioka Y, Takahashi A,
Takahashi T, Taniguchi T, Ishikawa Y and Yokoyama M: Trichostatin
A, an inhibitor of histone deacetylase, inhibits smooth muscle cell
proliferation via induction of p21(WAF1). J Atheroscler Thromb.
13:183–191. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Del Bufalo A, Bernad J, Dardenne C, Verda
D, Meunier JR, Rousset F, Martinozzi-Teissier S and Pipy B: Contact
sensitizers modulate the arachidonic acid metabolism of
PMA-differentiated U-937 monocytic cells activated by LPS. Toxicol
Appl Pharmacol. 256:35–43. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real -time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Pollack BP, Sapkota B and Boss JM:
Ultraviolet radiation-induced transcription is associated with
gene-specific histone acetylation. Photochem Photobiol. 85:652–662.
2009. View Article : Google Scholar
|
|
22
|
Kininis M, Chen BS, Diehl AG, Isaacs GD,
Zhang T, Siepel AC, Clark AG and Kraus WL: Genomic analyses of
transcription factor binding, histone acetylation and gene
expression reveal mechanistically distinct classes of
estrogen-regulated promoters. Mol Cell Biol. 27:5090–5104. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Aung HT, Schroder K, Himes SR, Brion K,
van Zuylen W, Trieu A, Suzuki H, Hayashizaki Y, Hume DA, Sweet MJ
and Ravasi T: LPS regulates proinflammatory gene expression in
macrophages by altering histone deacetylase expression. FASEB J.
20:1315–1327. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Strahl BD and Allis CD: The language of
covalent histone modifications. Nature. 403:41–45. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Natsume-Kitatani Y, Shiga M and Mamitsuka
H: Genome-wide integration on transcription factors, histone
acetylation and gene expression reveals genes co-regulated by
histone modification patterns. PLoS One. 6:e222812011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Balasubramani A, Winstead CJ, Turner H,
Janowski KM, Harbour SN, Shibata Y, Crawford GE, Hatton RD and
Weaver CT: Deletion of a conserved cis-element in the Ifng locus
highlights the role of acute histone acetylation in modulating
inducible gene transcription. PLoS Genet. 10:e10039692014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chung S, Sundar IK, Hwang JW, Yull FE,
Blackwell TS, Kinnula VL, Bulger M, Yao H and Rahman I: NF-κB
inducing kinase, NIK mediates cigarette smoke/TNFα-induced histone
acetylation and inflammation through differential activation of
IKKs. PLoS One. 6:e234882011. View Article : Google Scholar
|
|
28
|
Chung S, Sundar IK, Yao H, Ho YS and
Rahman I: Glutaredoxin 1 regulates cigarette smoke-mediated lung
inflammation through differential modulation of I{kappa}B kinases
in mice: Impact on histone acetylation. Am J Physiol Lung Cell Mol
Physiol. 299:L192–L203. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
de Ruijter AJ, van Gennip AH, Caron HN,
Kemp S and van Kuilenburg AB: Histone deacetylases (HDACs):
Characterization of the classical HDAC family. Biochem J.
370:737–749. 2003. View Article : Google Scholar
|
|
30
|
Khan N, Jeffers M, Kumar S, Hackett C,
Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, et al:
Determination of the class and isoform selectivity of
small-molecule histone deacetylase inhibitors. Biochem J.
409:581–589. 2008. View Article : Google Scholar
|