Introduction
X-linked ichthyosis (XLI, OMIM#308100) is an
X-linked recessive skin disorder that is caused by steroid
sulfatase (STS) deficiency (1).
The incidence rate of XLI is 1 in 6,000 males studied in various
populations (1). It is
characterized by desquamation of large brown scales on the limb and
trunk (2). Extracutaneous
manifestations, particularly corneal opacities and cryptorchidism,
are frequently observed (3). The
disease is caused by mutations in the STS gene that is
located on the short arm of the X-chromosome (Xp22.3) (4). Approximately 90% of patients with XLI
have large deletions involving the entire STS gene and
flanking regions (5,6).
Crigler-Najjar syndrome type I (CN-I, MIM#218800) is
a rare autosomal recessive disease caused by the genetic
alterations in the UPD-glucuronosyltransferase 1 family,
polypeptide A1 (UGT1A1) gene (7). It is characterized by high levels of
serum total bilirubin (342–684 µmol/l) caused by the
complete absence of the UGT1A1 enzyme, with a frequency of less
than one per million live births (8). The accumulation of unconjugated
bilirubin in the serum may cause irreversible neurological damage
(kernicterus). The brain damage can be prevented by intensive
phototherapy that is effective at least until adolescence (9). However, CN-I is a lethal disease when
not treated adequately and phenobarbital treatment is not effective
for the treatment of symptoms. Liver transplantation is the only
definitive treatment for CN-I. To date, the majority of
UGT1A1 mutations are single nucleotide substitutions,
including missense mutations and nonsense mutations. Large
deletions or insertions of UGT1A1 gene are less
reported.
Human genetic diseases are predominantly associated
with DNA variations, ranging from single nucleotide changes to
large DNA fragment variations, such as copy number variations
(CNVs). XLI and CN-I are two genetic diseases with Mendelian
inheritance patterns, however, they have distinct pathogenic
mechanisms and clinical manifestations. XLI predominantly arises
from CNVs involving the STS gene on the Xp22.3 region. By
contrast, CN-I is usually the result of point mutations in the
UGT1A1 gene, and CNVs involving this gene have rarely been
reported. Moreover, a coexistence of the two genetic alterations
has not been reported.
The present study presents a case of a Chinese male
patient affected by recessive XLI and CN-I, which were confirmed by
DNA quantitative polymerase chain reaction (PCR) and single
nucleotide polymorphism (SNP) array analysis. To the best of our
knowledge, this is the first report this co-morbidity.
Materials and methods
DNA samples
Genomic DNA samples were extracted from the
peripheral blood leukocytes of the patient and his parents, using
the phenol-chloroform method (10). Informed consent was obtained from
the patient's parents. This project was approved by the ethics
committee of the Capital Institute of Pediatrics (Beijing,
China).
Quantitative PCR for STS gene-copy number
analysis
As 90% of the patients with XLI have large deletions
involving the entire STS gene, the copy numbers of this gene
were directly detected by quantitative PCR on genomic DNA. Primers
were designed using the Primer Express 3.0 software (Applied
Biosystems, Thermo Fisher Scientific, Inc., Waltham, MA, USA). The
STS primers used were as follows: STS-E1F,
5′-GTTGCCCCTTCTGAAGAATCC-3′; STS-E1R, 5′-GAGGCGGAGACACTC TTTGC-3′;
STS-E10F, 5′-TGGAGTGCAGTGGTGCAATC-3′; STS-E10R,
5′-GTGACTTGGGAGGCTGAAGTG-3′. The quantitative PCR was conducted
using the ABI Prism 7500 sequence detection system (Applied
Biosystems, Thermo Fisher Scientific Inc.) as described by Wilke
et al (11). Data were
analyzed using the ABI 7500 Prism sequence detection software
(Applied Biosystems, Thermo Fisher Scientific, Inc.).
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as
an internal control gene for gene copy number analysis. The
2−ΔΔCq method for relative quantification was used to
evaluate STS gene copy numbers of the patient and his
parents in comparison with those of healthy controls. DNA from
healthy controls served as a calibrator that had a
2−ΔΔCq of 1. The 2−ΔΔCq value of the
inspected sample is the ratio of the target gene copy number of
inspected sample to the calibrator.
SNP array analysis
A genome-wide SNP array, Human CytoSNP-12 DNA
Analysis BeadChip kit (Illumina, Inc., San Diego, CA, USA) was used
to examine the deletion sizes of the two regions (2q37.1 and
Xp22.31) containing STS and UGT1A1 genes. The
genome-wide SNP array was composed of 300,000 probe markers.
Briefly, 4 µl DNA sample (200 ng) was mixed with 4 µl
NaOH (0.1 mol/l) solution, followed by a 10-min incubation at room
temperature for DNA denaturation. Next, 68 µl MA2 solution
and 75 µl MSM solution (both from Illumina, Inc.) were added
to the mixture to allow DNA renaturation. The reaction solution was
put in the 37°C hybrid furnace for 20 h. The resulting DNA
fragments were resuspended in RA1 solution (Illumina, Inc.). and
the DNA solution was loaded onto the chip. The chip was kept in an
airtight hybrid box (Illumina, Inc.) to permit hybridization for 16
h at 48°C. When the chip was washed to remove non-hybridized and
nonspecific hybridized DNA fragments, single base extension and
chip staining reactions were initiated. The iScan scanning system
(Illumina, Inc.) was used to collect data, and the data were
further analyzed by Karyostudio software v1.4 (Illumina, Inc.).
Results
Clinical presentation
The male patient is the first offspring of healthy
and nonconsanguineous parents. He was born at term by normal
delivery; however, his mother had the premonitory symptoms of
miscarriage two months into the pregnancy. The patient presented
with yellowing of the skin 6 days after birth. He was admitted to
the Capital Institute of Pediatrics due to yellowing of the skin
that did not reduce at 4.5 months old. His total serum bilirubin
level was 520.6 µmol/l and serum unconjugated bilirubin
level was 499.6 µmol/l. The patient succumbed to kernicterus
at 13 months of age, due to molecularly confirmed CN-I syndrome
(12). However, upon reviewing the
related medical records, other abnormal manifestations in the
patient that could not be fully attributed to CN-I alone, were
identified. Upon admission, the physical examination indicated that
the patient was 7,000 g (20th) in weight and 66 cm in length
(50–75th), with a head circumference of 41 cm (20th), which
revealed that the patient had growth retardation. He could not
raise his head at 4 months old of age, which suggested slight
retardation of mental and motor development. A dispersed congestive
rash were observed on his face and head, and rhombus-shaped brown
pigmentation and furfuration were observed on his trunk and four
limbs. A follow-up survey with the family revealed that the
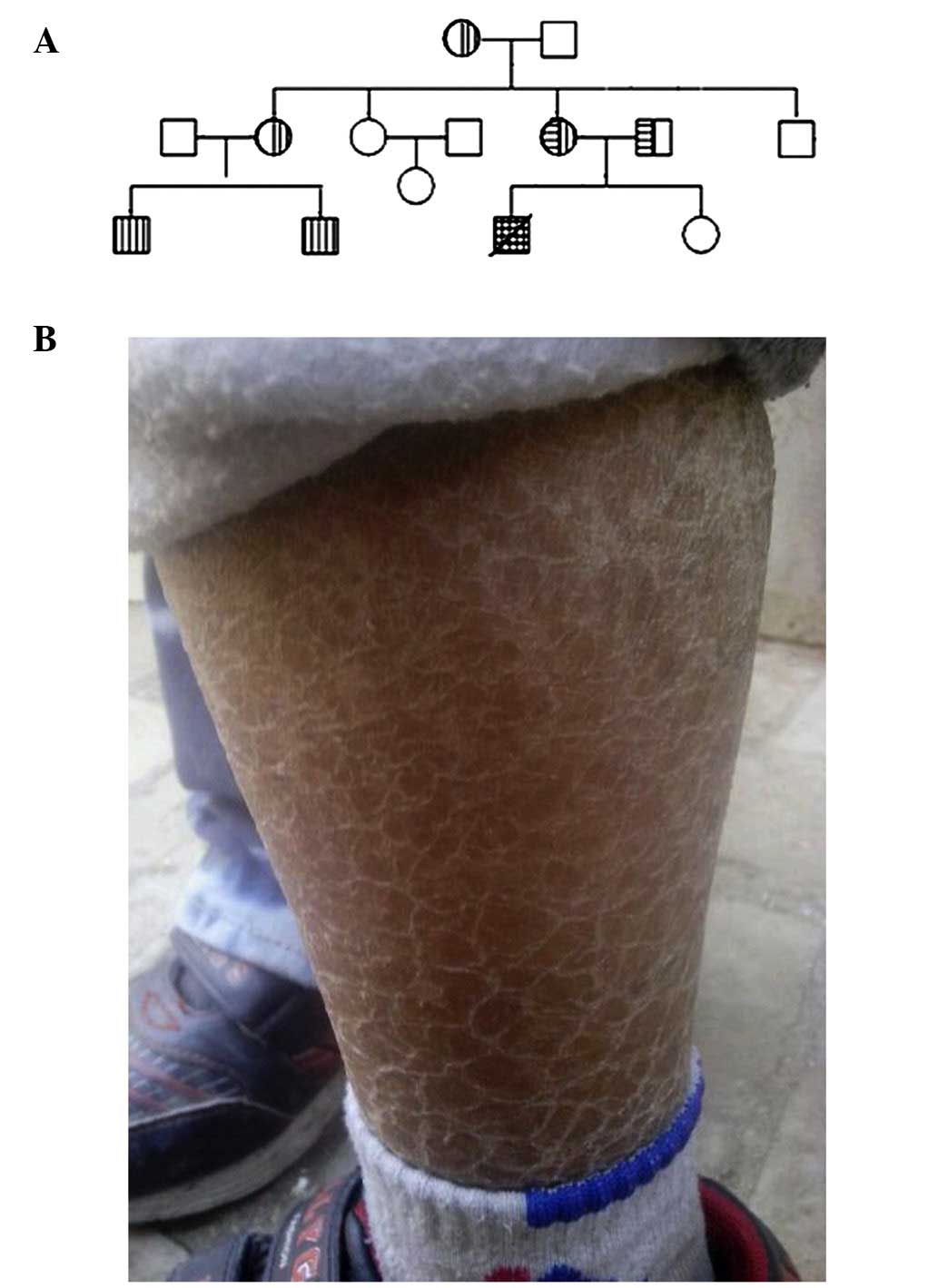
patient's two cousins also suffered from ichthyosis (Fig. 1); thus, XLI was suspected. Apart
from ichthyosis, the patient had inspiratory stridor after birth
and was diagnosed with congenital laryngeal cartilage softening
disease. Abdominal computer tomography scanning indicated that the
patient had left renal dysplasia and left renal artery stenosis.
However, his renal function and routine urine test results were
normal.
Gene copy number changes
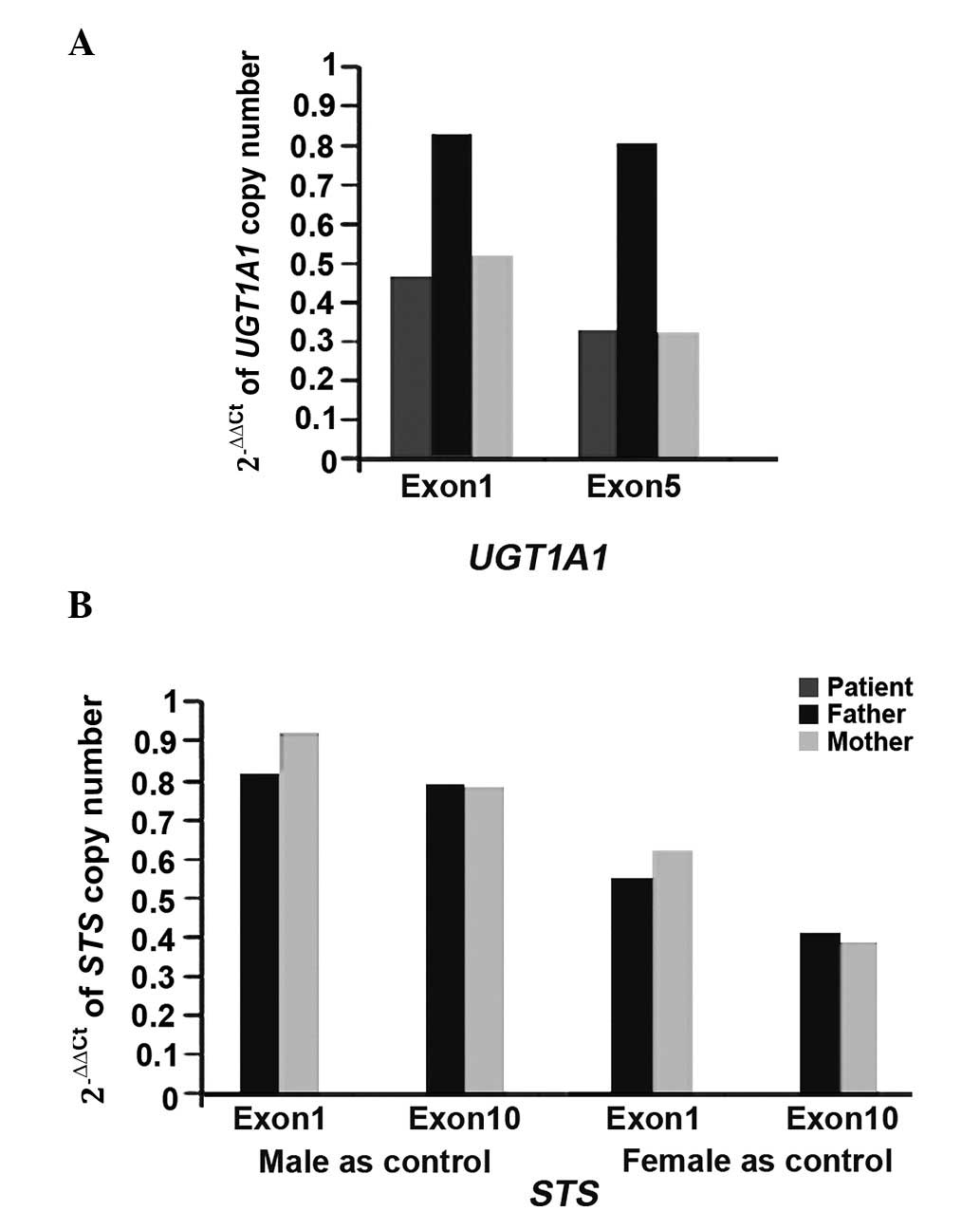
For the STS gene, the patient's mother and father
both carried only one copy number (N=1), although the male patient
did not carry the gene (N=0). The results of real-time quantitative
PCR are shown in Fig. 2, and the
2−ΔΔCq value of the control (male or female) was
considered to be 1.
The 'homozygous mutation' of c.1253delT in exon 4 of
the UGT1A1 gene was detected in the patient by DNA
sequencing in our previous study (12). The same mutation was observed in
his father, but not in his mother. It is possible that an allele
lacking the entire UGT1A1 gene may be transmitted to the
patient from the mother who had a single UGT1A1 gene copy.
Therefore, for the patient with one point mutation (c.1253delT) on
one allele and with an entire deletion of the UGT1A1 gene on
the other allele, the possibility existed of falling into the
'trap' of 'homozygous mutation' (c.1253delT) in the patient by
direct DNA sequencing. Thus, quantitative PCR was performed to
detect the copy number of UGT1A1 gene in the core members.
The results demonstrated that his father carried two copies of the
UGT1A1 gene, with c.1253delT detected on one of alleles. His
mother only had one copy of the UGT1A1 gene without base
changes. The patient also carried a single copy of the
UGT1A1 gene that harbored the c.1253delT mutation.
Deletion range detection
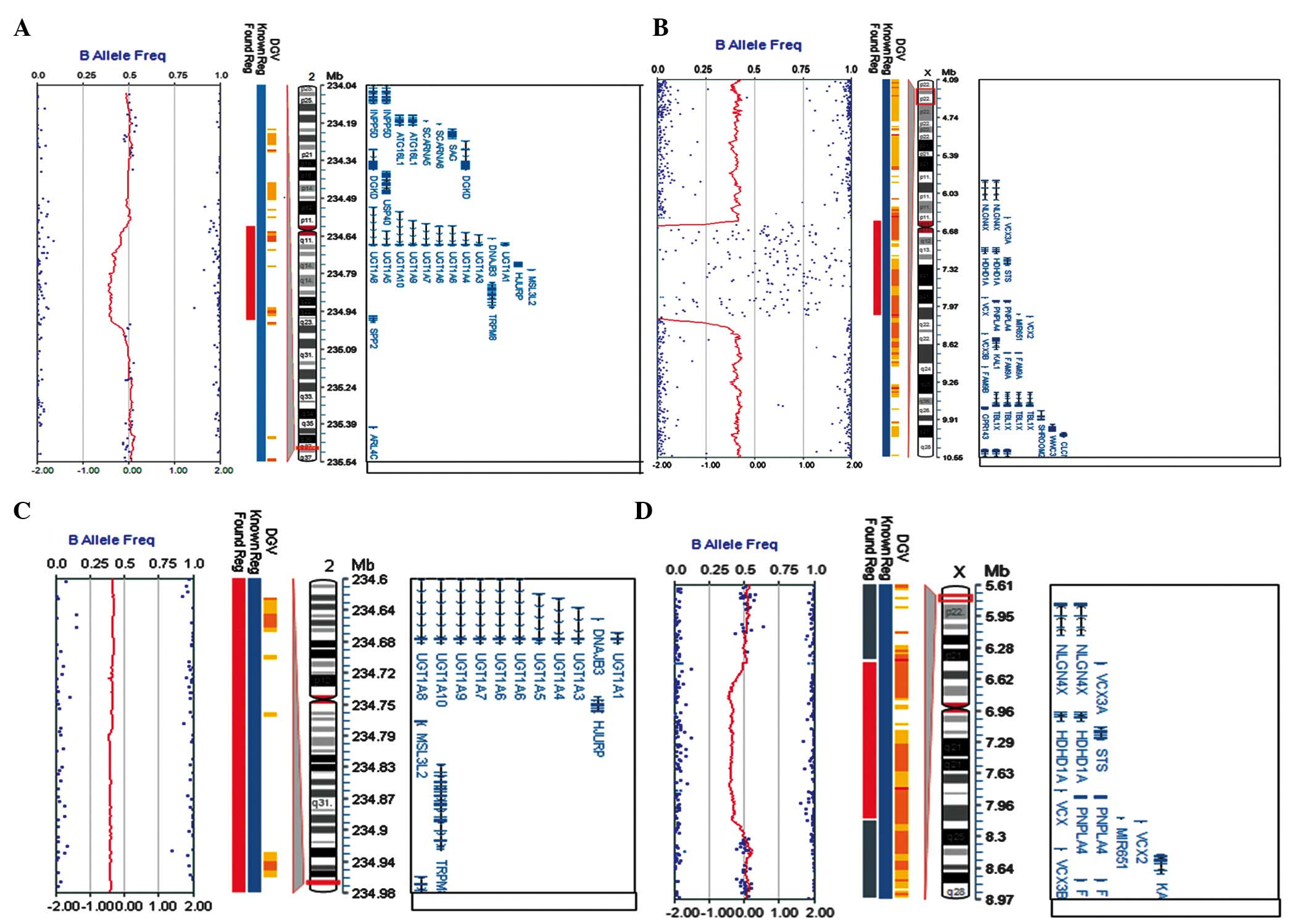
SNP array results are shown in Fig. 3. Two CNVs were confirmed for this
patient: One maternal microdeletion was located on ChrXp22.31 with
an estimated size of 1.61 M; the other maternal microdeletion was
found on Chr2q37.1 (Table I), with
an estimated size of 374-kb (Fig. 3A
and B). Moreover, SNP array results showed that his mother had
the same microdeletion of 374 kb on Chr2q37.1, but a larger
deletion (~1.67 Mb) on ChrXp22.31, including STS and
additional five genes (for example, mental retardation related gene
VCX3A) (Fig. 3C and D).
Thus, these data suggested that the two CNVs in the patient may be
inherited from his mother. One CNV contained an XLI-causing gene,
STS, and the other CNV comprised a causative gene for CN-I,
UGT1A1. These genetic abnormalities were well matched to the
main clinical phenotypes of the patient, i.e, ichthyosis and
icterus (Table I).
 | Table IDeletion result of single nucleotide
polymorphism array analysis of the patient. |
Table I
Deletion result of single nucleotide
polymorphism array analysis of the patient.
| Region | From | To | Length (bp) | Gene | Related
phenotype |
|---|
| Chr2p37.1 | 234604215 | 234978363 | 374148 | UGT1A8;
UGT1A10; UGT1A9; UGT1A7; UGT1A6;
UGT1A5; UGT1A4; UGT1A3; DNAJB3;
UGT1A1; HJURP; MSL3L2; TRPM8 | Crigler-Najjar
syndrome |
| ChrXp22.31 | 6516735 | 8131442 | 1614707 | HDHD1A;
STS; VCX; PNPLA4; MIR651 | Ichthyosis |
Discussion
In this study, a unique patient with XLI and CN-I,
two recessive inherited diseases, was described. The two diagnoses
were confirmed by molecular genetic evidence, as the patient
exhibited a maternal 1.61 Mb deletion involving the STS
gene, and a 374 Kb maternal deletion involving the UGT1A1
gene combined with a paternal UGT1A1 gene mutation
(c.1253delT). Since these two recessive inherited diseases existed
simultaneously in the same patient but independently with each
other, it was speculated that the co-occurrence of two conditions
was incidental. To the best of our knowledge, this is the first
reported case with this comorbidity.
The patient's clinical symptoms of the skin were
consistent with the diagnosis of ichthyosis. Pedigree analysis
showed that there were two affected cousins in the extended family,
which was consistent with the transmission characteristics of
X-linked recessive diseases. Up to 90% of XLI patients have large
gene fragment deletions involving the entire STS gene and
its flanking regions, while the minorities of cases display partial
deletions or point mutations in the gene (5,6,13).
In this study, quantitative PCR and SNP microarray analysis
confirmed the deletion of the entire STS gene in this
patient. A previous study demonstrated that this aberration was the
result of the unequal crossing-over in the two regions between the
highly conserved CRI-S232 repeat regions flanking the STS
gene (14). The deletion size of
XLI in this patient was 1.61 Mb, which was close to the common
1.5-Mb deletion. Recent studies have shown that nervous system
abnormalities, short stature, mental retardation, Kallmann syndrome
or other clinical features may occur, when the deletion size on
Xp22.3 is >1.5 Mb (15,16). Accordingly, the VCX3A gene
located ~0.7 Mb telomeric to the STS gene has been proposed
as the candidate mental retardation gene on Xp22.3 (17,18).
In the present study, the STS and VCX3A genes were
deleted on one of the X chromosomes of the patient's mother who had
a normal phenotype. By contrast, the male patient presented with
slight mental retardation, although he only lost the STS
gene, but not the VCX3A gene. Such a disconnection of
genotype and phenotype was considered to be due to the fact that
number and location of the probes and hybridization signals of this
region may limit the capacity to determine the accurate deletion
size by SNP analysis. Unfortunately, we failed to repeat the SNP
analysis or conduct another experiment to confirm the results, due
to the lack of availability of the patient DNA samples. In
addition, although the deletion range of the patient did not
include the Kallmann syndrome 1 protein, certain clinical
manifestations of Kallmann syndrome were observed, such as left
renal dysplasia. Conversely, he did not have the most common
symptoms of XLI (corneal opacity or cryptorchidism). Moreover, the
pathogenic mechanism for other complex manifestations, such as
congenital laryngeal cartilage softening disease, require further
elucidation.
To date, >130 UGT1A1 gene mutations have
been identified, including 91 single nucleotide changes (77
missense mutations and 14 nonsense mutations), 21 deletions, 10
insertions and 8 mutations located in the promoter and introns
(19). Complete loss of the
UGT1A1 gene is rarely reported. The present study initially
performed direct DNA sequencing to screen the mutations, based on
the feature of the UGT1A1 gene mutation spectrum. This
result revealed that the patient had a homozygous mutation-c.1253
Del T of the UGT1A1 gene, which could not be fully explained
by mutation transmission analysis of his parents as the same
mutation was only observed in his father and not in his mother.
Thus, this phenomenon prompted us to further careful analysis of
DNA sequencing data for UGT1A1, the causative gene of the
monogenic disease. Since the parents of the patient were
unconsanguineous, it was presumed that the patient's mother only
carried one copy of normal UGT1A1 gene as indicated by DNA
sequence analysis. Subsequently, PCR and SNP microarray analysis
confirmed that the mother only had a single copy of UGT1A1
gene. It is extremely rare for CN-I to be caused by a point
mutation and a complete deletion of the UGT1A1 gene. As the
clinical phenotypes of this patient were more complex than what the
known genotypes could explain, it was necessary to analyze the
deletion range to understand the correlation between genotype and
phenotype. Therefore, a Genome-wide SNP array analysis was
conducted in this study.
CNV is a form of DNA structural variation (≥1 kb),
which is present at a variable copy number, when compared with a
reference genome. It occurs by recombination-based and
replication-based mechanisms (18). Although CNVs can represent benign
polymorphic variants widely distributed in the human genome, there
are numerous CNVs that can cause Mendelian or sporadic traits, or
be associated with complex diseases (20). To assess the pathogenicity of CNVs,
the ISCA Consortium has published the criteria in their consensus
statement of 2010 (21). For
example, if the CNVs contain morbid MIM genes, they are probably
pathogenic. By contrast, when the CNVs are inherited from a healthy
parent, they are probably benign (22). In this study, the proband has been
confirmed to carry two CNVs: One was a 374 Kb microdeletion located
on Chr2q37.1 and the other was ~1.61 M deletion located on
ChrXp22.31. The two CNVs contained two disease-causing genes,
UGT1A1 and STS, respectively. Family pedigree
analysis showed that the two CNVs were inherited from the patient's
mother. It was assumed that the 1.61 M deletion may be transmitted
from the proband's grandmother as both cousins exhibited the
phenotype of ichthyosis. Furthermore, a 374 kb microdeletion that
resulted in CN-I was passed from the proband's mother. However, it
could not be determined whether this microdeletion was inherited
from the grandparents or was de novo in his mother. Cases
with the two recessive genetic diseases have not been reported. The
CN-I of the patient in the present study resulted from a rare 374
Kb maternal microdeletion combined with a paternal c.1253 delT
mutation. Coincidentally, the proband was a male and also carried
the maternal 1.61 M deletion located on ChrXp22.31, which caused
him to lose the STS gene and consequentially develop
XLI.
In conclusion, this study suggested that rare
diseases may not simply be associated with the affected gene but
may also be concurrent with genomic variations, such as CNVs. It
also highlighted the importance of conducting detailed clinical
examinations and unbiased genetic analyses, particularly for the
patients presenting with clinical phenotypes that could not be
fully explained by general genetic testing results. Further studies
using genome-wide scanning techniques (e.g., SNP array) may aid in
determining disease-causing genetic alterations to understand the
phenotypes in the affected individuals. Gaining a full
understanding of the phenotype may assist in enabling a more
accurate diagnosis to be made, and also in providing an improved
basis for rational treatment strategies.
Acknowledgments
The authors would like to thank the family members
for participating. They would also like to acknowledge Dr Gao Ying
(Department of Dermatology, Capital Institute of Pediatrics) for
providing helpful comments.
Abbreviations:
|
CN-I
|
Crigler-Najjar syndrome
|
|
UGT1A1 gene
|
UPD-glucuronosyltransferase 1 family,
polypeptide A1 gene
|
|
XLI
|
X-linked ichthyosis
|
|
STS
|
steroid sulfatase
|
|
CNVs
|
copy number variations
|
|
SNP
|
single nucleotide polymorphism
|
References
|
1
|
Shapiro LJ, Weiss R, Webster D and France
JT: X-linked ichthyosis due to steroid-sulphatase deficiency.
Lancet. 14:70–72. 1978. View Article : Google Scholar
|
|
2
|
Hosomi N, Oiso N, Fukai K, Hanada K,
Fujita H and Ishii M: Deletion of distal promoter of VCXA in a
patient with X-linked ichthyosis associated with borderline mental
retardation. J Dermatol Sci. 45:31–36. 2007. View Article : Google Scholar
|
|
3
|
Traupe H and Ropers HH: Cryptorchidism and
hypogenitalism in X-linked recessive ichthyosis vulgaris. Hum
Genet. 60(206)1982. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tiepolo L, Zuffardi O, Fraccaro M, di
Natale D, Gargantini L, Müller CR and Ropers HH: Assignment by
deletion mapping of the steroid sulfatase X-linked ichthyosis locus
to Xp223. Hum Genet. 54:205–206. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yen PH, Allen E, Marsh B, Mohandas T, Wang
N, Taggart RT and Shapiro LJ: Cloning and expression of steroid
sulfatase cDNA and the frequent occurrence of deletions in STS
deficiency: Implications for X-Y interchange. Cell. 49:443–454.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bonifas JM, Morley BJ, Oakey RE, Kan YW
and Epstein EH Jr: Cloning of a cDNA for steroid sulfatase:
Frequent occurrence of gene deletions in patients with recessive X
chromosome-linked ichthyosis. Proc Natl Acad Sci USA. 84:9248–9251.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Crigler JF Jr and Najjar VA: Congenital
familial nonhemolytic jaundice with kernicterus. Pediatrics.
10:169–180. 1952.PubMed/NCBI
|
|
8
|
Petit FM, Gajdos V, Francoual J, Capel L,
Parisot F, Poüs C and Labrune P: Allelic heterogeneity of
Crigler-Najjar type I syndrome: A study of 24 cases. Clin Genet.
66:571–572. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Van der Veere CN, Sinaasappel M, McDonagh
AF, Rosenthal P, Labrune P, Odièvre M, Fevery J, Otte JB, McClean
P, Bürk G, et al: Current therapy for Crigler-Najjar syndrome type
1: Report of a world registry. Hepatology. 24:311–315. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Santos EM, Paula JF, Motta PM, Heinemann
MB, Leite RC, Haddad JP, Del Puerto HL and Reis JK: Comparison of
three methods of DNA extraction from peripheral blood mononuclear
cells and lung fragments of equines. Genet Mol Res. 9:1591–1598.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wilke K, Duman B and Horst J: Diagnosis of
haploidy and triploidy based on measurement of gene copy number by
real-time PCR. Hum Mutat. 16:431–436. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li Y, Qu YJ, Zhong XM, Cao YY, Jin LM, Bai
JL, Ma X, Jin YW, Wang H, Zhang YL and Song F: Two unrelated
patients with rare Crigler-Najjar syndrome type I: Two novel
mutations and a patient with loss of heterozygosity of UGT1A1 gene.
J Zhejiang Univ Sci B. 15:474–481. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Murtaza G, Siddiq S, Khan S, Hussain S and
Naeem M: Molecular study of X-linked ichthyosis: Report of a novel
2-bp insertion mutation in the STS and a very rare case of
homozygous female patient. J Dermatol Sci. 74:165–167. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fukami M, Kirsch S, Schiller S, Richter A,
Benes V, Franco B, Muroya K, Rao E, Merker S, Niesler B, et al: A
member of a gene family on Xp22.3, VCX-A, is deleted in patients
with X-linked nonspecific mental retardation. Am J Hum Genet.
67:563–573. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Paige DG, Emilion GG, Bouloux PM and
Harper JI: A clinical and genetic study of X-linked recessive
ichthyosis and contiguous gene defects. Br J Dermatol. 131:622–629.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Van Steensel MA, Vreeburg M, Engelen J,
Ghesquiere S, Stegmann AP, Herbergs J, van Lent J, Smeets B and
Vles JH: Contiguous gene syndrome due to a maternally inherited
8.41 Mb distal deletion of chromosome band Xp22.3 in a boy with
short stature, ichthyosis, epilepsy, mental retardation, cerebral
cortical heterotopias and Dandy-Walker malformation. Am J Med Genet
A. 146:2944–2949. 2008. View Article : Google Scholar
|
|
17
|
Gohlke BC, Haug K, Fukami M, Friedl W,
Noeker M, Rappold GA and Haverkamp F: Interstitial deletion in
Xp22.3 is associated with X linked ichthyosis, mental retardation
and epilepsy. J Med Genet. 37:600–602. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lahn BT and Page DC: A human
sex-chromosomal gene family expressed in male germ cells and
encoding variably charged proteins. Hum Mol Genet. 22:311–319.
2000. View Article : Google Scholar
|
|
19
|
Canu G, Minucci A, Zuppi C and Capoluongo
E: Gilbert and Crigler Najjar syndromes: An update of the
UDP-glucuronosyltransferase 1A1 (UGT1A1) gene mutation database.
Blood Cells Mol Dis. 50:273–280. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang F, Gu W, Hurles ME and Lupski JR:
Copy number variation in human health, disease and evolution. Annu
Rev Genomics Hum Genet. 10:451–481. 2009. View Article : Google Scholar
|
|
21
|
Miller DT, Adam MP, Aradhya S, Biesecker
LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE,
Epstein CJ, et al: Consensus statement: Chromosomal microarray is a
first-tier clinical diagnostic test for individuals with
developmental disabilities or congenital anomalies. Am J Hum Genet.
14:749–764. 2010. View Article : Google Scholar
|
|
22
|
Schaaf CP, Wiszniewska J and Beaudet AL:
Copy number and SNP arrays in clinical diagnostics. Annu Rev
Genomics Hum Genet. 12:25–51. 2011. View Article : Google Scholar : PubMed/NCBI
|