Introduction
Endothelial progenitor cells (EPCs) can
differentiate into endothelial cells (ECs) and are important in
angiogenesis (1). It has been
demonstrated that oxidative stress-induced endothelial dysfunction
can cause ischemia/reperfusion injury, and the proliferation and
differentiation of EPCs are essential for the treatment of this
type of injury. Hypoxia has been demonstrated to mobilize EPCs from
the bone marrow into the peripheral blood (2), and induce ECs to secret chemokines
for the recruitment and migration of EPCs to hypoxic tissues
(3). In addition, hypoxia can
induce the expression of hypoxia-inducible factor-1α (HIF-1α), an
important transcriptional factor that is involved in the
proliferation and differentiation of EPCs (4).
Apelin is an endogenous ligand for the G
protein-coupled receptor, APLNR (5). It has been suggested that
Apelin/APLNR signaling can be activated by HIF-1α in hypoxia
neonatal cardiomyocytes (6). In
addition, downregulated serum levels of Apelin are associated with
EPC mobilization induced by acute myocardial infarction (7) and Apelin deficiency impairs EPC
sprouting in ischemia-reperfusion injury (8). These findings suggest that Apelin may
be involved in the regulation of EPC mobilization and sprouting.
However, whether the Apelin/APLNR signaling is involved in
hypoxia-induced EPC proliferation has not been investigated
previously.
The mitogen-activated protein kinase (MAPK)
signaling pathway is important in the regulation of cellular
survival, proliferation, differentiation and migration (9). It has been demonstrated that the MAPK
signaling pathway is involved in the regulation of EPC
proliferation, for example, the effect of granulocyte macrophage
colony-stimulating factor on the modulation of EPC proliferation is
mediated by the MAPK signaling pathway (10). Our previous study revealed that
Apelin/APLNR signaling promotes hypoxia-induced proliferation of
EPCs via phosphoinositide 3 kinase/Akt signaling (11). However, whether Apelin/APLNR
signaling regulates EPC proliferation via MAPK signaling remains to
be elucidated
The aim of the present study was to determine the
underlying molecular mechanisms by which Apelin/APLNR signaling
regulates the hypoxia-induced proliferation of EPCs.
Materials and methods
Cell isolation and culture
EPCs were isolated from the umbilical cord at the
Second Xiangya Hospital of Central South University in September
2012. Briefly, umbilical cord tissue sample was diluted with
Dulbecco's phosphate-buffered saline at a ratio of 1:1 and overlaid
onto 1.077 g/ml Ficoll-Paque™ Premium (GE Healthcare Bio-Sciences,
Pittsburgh, PA, USA) prior to centrifugation for 30 min at 400 × g.
The monocytes were collected and seeded into tissue culture plates
coated with fibronectin (EMD Millipore, Billerica, MA, USA), and
cultured in endothelial growth medium (EGM)-2 (Thermo Fisher
Scientific, Waltham, MA, USA) supplemented with 20% fetal bovine
serum (FBS; Tianhang Biological Technology Co., Ltd., Hangzhou,
China) at 37°C in a humidified incubator containing 5%
CO2. The culture media was changed every two days until
EPC colonies appeared. Typical colonies appear between days 5 and
10, and were passaged at sub-confluence. The present study was
approved by the ethics committee of the Second Xiangya Hospital of
Central South University (Changsha, China).
Hypoxia treatment of the EPCs
Prior to hypoxia treatment, the EPCs
(5×105) were cultured in serum-free EGM-2 at 37°C, 5%
CO2 for 12 h. Subsequently, the EPCs were cultured in
EGM-2 containing 20% FBS at 37, 1% O2/94%
N2/5% CO2 for 6, 12, 24 and 48 h.
Cell proliferation assay
An MTT assay was performed to determine the cell
proliferation. In brief, 10 mg/ml MTT (Thermo Fisher Scientific)
was added to the medium following culture. Following incubation for
4 h, the reaction was terminated by removal of the supernatant and
the addition of 100 µl dimethyl sulfoxide (DMSO; Thermo
Fisher Scientific) to dissolve the formazan product. After 30 min,
the optical density (OD) of each well was measured at 570 nm using
a microplate reader (ELx808; Bio-Tek Instruments, City, ST,
USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
TRIzol reagent (Life Technologies, Carlsbad, CA,
USA) was used to extract the total RNA from the EPCs, according to
the manufacturer's instructions. The total RNA was reverse
transcribed into cDNA using a RevertAid First-Strand cDNA Synthesis
kit (Thermo Fisher Scientific), according to the manufacturer's
instructions. The mRNA expression level was determined using iQTM
SYBR Green Supermix (Bio-Rad Laboratories, Inc., Hercules, CA,
USA), according to the manufacture's instructions. The reaction
mixture contained 1 µl cDNA solution, 10 µl PCR
master mix, 2 µl primers (Sangon Biotech Co., Ltd.,
Shanghai, China), and 7 µl H2O were mixed to
obtain a final reaction volume of 20 µl. The qPCR was
conducted using an ABI7500 PCR system (Invitrogen; Thermo Fisher
Scientific, Inc.) with the following reaction conditions:
Pre-degeneration at 95°C for 1 min, followed by 40 cycles of 95°C
for 15 sec, 60°C for 15 sec and 72°C for 1 min. The specific primer
pairs used were as follows: HIF-1α, sense
5′-GAACGTCGAAAAGAAAAGTCTCG-3′ and antisense
5′-CCTTATCAAGATGCGAACTCACA-3′; Apelin, sense
5′-GTCTCCTCCATAGATTGGTCTGC-3′ and antisense
5′-GGAATCATCCAAACTACAGCCAG-3′; APLNR, sense
5′-CTCTGGACCGTGTTTCGGAG-3′ and antisense
5′-GGTACGTGTAGGTAGCCCACA-3′; and GAPDH (as an inter nal reference)
sense 5′-GGAGCGAGATCCCTCCAAAAT-3′ and antisense
5′-GGCTGTTGTCATACTTCTCATGG-3′. Independent experiments were
repeated three times. The relative expression levels of mRNA were
analyzed using the 2−∆∆Ct method (12).
Transfection
Transfection of the cells was performed using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's instructions. For functional
analysis, the EPCs were transfected with either an Apelin plasmid,
Apelin-specific siRNA, an APLNR plasmid or APLNR-specific siRNA
(Nlunbio, Changsha, China).
Western blot analysis
The cells were solubilized in cold
radioimmunoprecipitation assay lysis buffer. The proteins were
quantified using a Bicinchoninic Acid Protein Assay kit (Thermo
Fisher Scientific, Inc.), according to the manufacturer's
instructions. The resulting proteins (50 µg) were separated
with 12% SDS-PAGE (Beyotime Institute of Biotechnology, Shanghai,
China) and transferred onto a polyvinylidene difluoride (PVDF)
membrane (Thermo Fisher Scientific), which was then incubated with
Tris-buffered saline with 1% Tween 20 (Thermo Fisher Scientific)
containing 5% skimmed milk at 4°C for overnight. Following
incubation, the PVDF membrane was incubated with mouse anti-HIF-α,
mouse anti-Apelin, mouse anti-APLNR and mouse anti-GAPDH primary
antibodies (Abcam, Cambridge, UK), respectively, at room
temperature for 3 h. Following washing with PBS with 1% Tween 20
(PBST; Thermo Fisher Scientific) three times, the PVDF membrane was
incubated with rabbit anti-mouse secondary antibody (Abcam) at room
temperature for 1 h. Following washing with PBST three times, an
ECL kit (Pierce Chemical, Rockford, IL, USA) was used to perform
chemiluminescent detection. The relative expression of proteins
were determined using Image-Pro plus software 6.0, presented as the
density ratio, compared with GAPDH.
Statistical analysis
All data are expressed as the mean ± standard
deviation of three independent experiments. Statistical analyses of
differences were performed using one-way analysis of variance with
SPSS 17 software (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
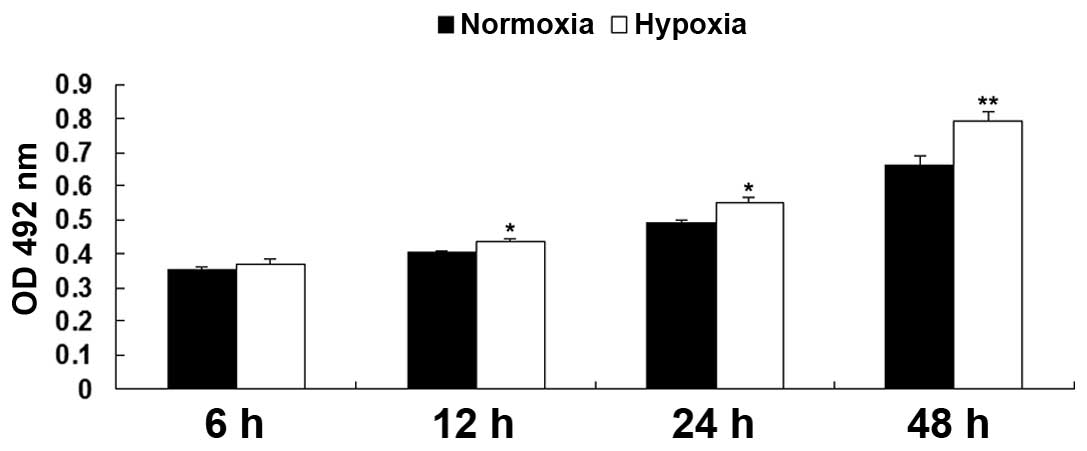
Hypoxia promotes EPC proliferation
The proliferation rates of the EPCs were examined
under hypoxia and normoxia using an MTT assay. As shown in Fig. 1, following culture in hypoxia for
6, 12, 24, and 48 h, the relative proliferation rates of the EPCs
were notably upregulated, compared with the EPCs cultured in
normoxia, indicating that hypoxia promoted EPC proliferation.
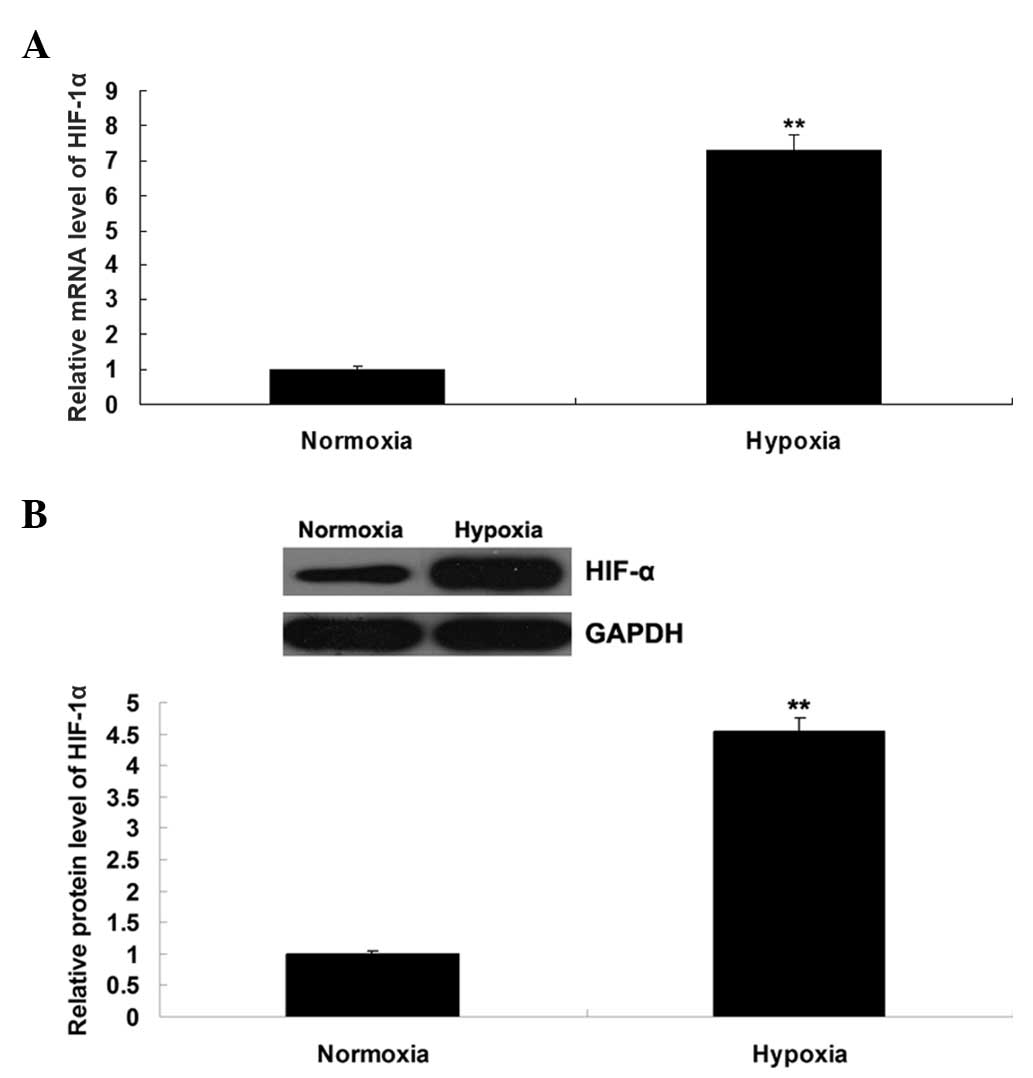
Hypoxia induces the upregulation of
HIF-1α and Apelin/APLNR signaling
The expression level of HIF-1α was further
determined by performing RT-qPCR and western blot assays. As shown
in Fig. 2A and B, the mRNA and
protein expression levels of HIF-1α were notably upregulated in the
EPCs cultured under hypoxia, compared with those cultured in
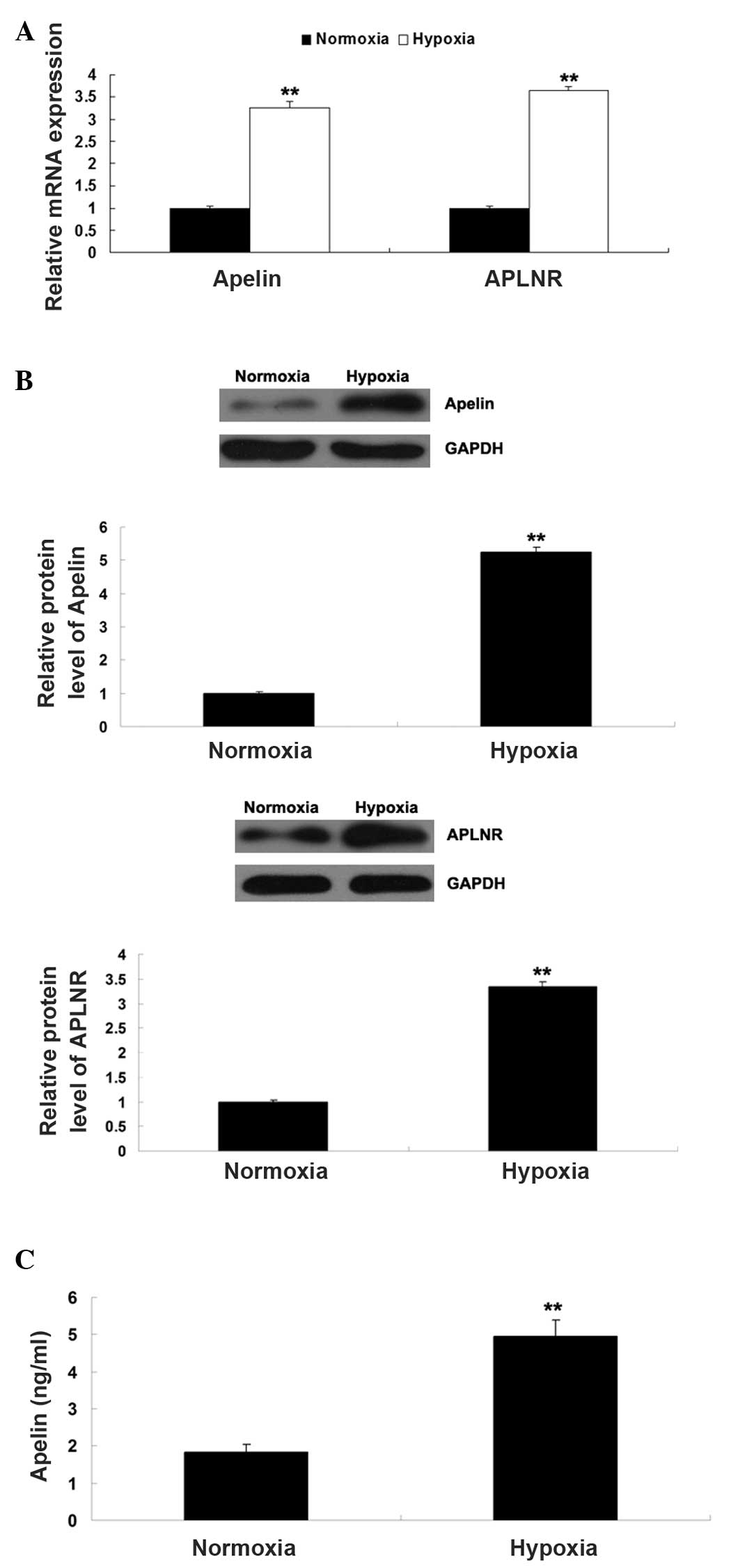
normoxia. As Apelin/APLNR signaling has been found to be regulated
by HIF-1α, the present study further determined the expression
levels of Apelin and APLNR in EPCs cultured under hypoxia or
normoxia, respectively. As shown in Fig. 3A and B, the mRNA and protein
expression levels of Apelin and APLNR were significantly
upregulated in the EPCs cultured under hypoxia, compared with those
cultured in normoxia. In addition, the secretion of Apelin was
determined using ELISA. As shown in Fig. 3C, the secretion level of Apelin was
also increased in the EPCs cultured under hypoxia, compared with
those cultured in normoxia. Taken together, these findings
suggested that hypoxia induced the upregulation of HIF-1α and its
downstream Apelin/APLNR signaling in the EPCs.
Role of Apelin/APLNR signaling in EPC
proliferation
The role of Apelin/APLNR signaling was further
investigated in the proliferation of EPCs cultured under hypoxia or
normoxia, respectively. The EPCs were initially transfected either
with an Apelin plasmid or siRNA, in order to upregulate or
down-regulate Apelin/APLNR signaling, respectively. As shown in
Fig. 4A and B, the mRNA and
protein expression levels of Apelin increased in the EPCs
transfected with the Apelin plasmid, and reduced in the EPCs
transfected with the Apelin siRNA. The present study subsequently
determined the proliferation of EPCs cultured under hypoxia or
normoxia using an MTT assay. As shown in Fig. 4C, the proliferation of the EPCs in
normoxia + Apelin group was higher than that in the normoxia group,
suggesting that the upregulation of Apelin enhanced EPC
proliferation. In addition, the proliferation of the EPCs in the
hypoxia + Apelin siRNA group was lower than that in the hypoxia
group, suggesting that the inhibition of Apelin suppressed
hypoxia-induced EPC proliferation.
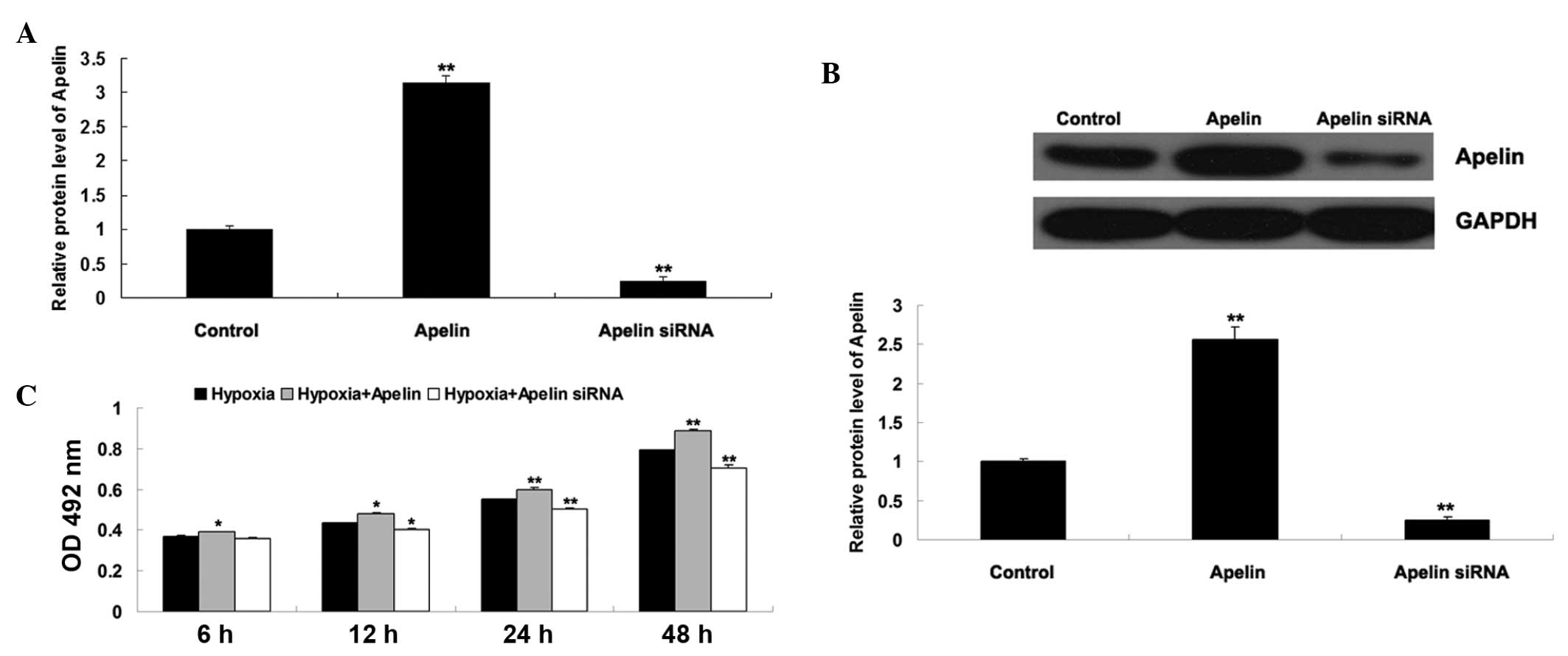 | Figure 4(A) Reverse transcription-quantitative
polymerase chain reaction was performed to examine the mRNA
expression of Apelin in EPCs transfected with either the Apelin
plasmid or Apelin siRNA. GAPDH was used as an internal reference.
Untransfected cells were used as a control. **P<0.01,
vs. control. (B) Western blotting was performed to examine the
protein expression of Apelin in EPCs transfected with either the
Apelin plasmid or Apelin siRNA. GAPDH was used as an internal
reference. **P<0.01, vs. control. (C) MTT assay was
performed to determine the cell proliferation rates of EPCs in each
group at 6 h, 12 h, 24 h and 48 h. Hypoxia: EPCs cultured under
hypoxia. Hypoxia + Apelin: EPCs transfected with the Apelin plasmid
and cultured under hypoxia. Hypoxia + Apelin siRNA: EPCs
transfected with Apelin siRNA and cultured under hypoxia.
*P<0.05, vs. hypoxia. **P<0.01, vs.
hypoxia. Data are expressed as the mean ± standard deviation. EPC,
endothelial progenitor cell; APLNR, Apelin receptor; OD, optical
density; siRNA, small interfering RNA. |
To further confirm the hypothesis that Apelin/APLNR
signaling is involved in EPC proliferation, the present study
transfected EPCs with either an APLNR plasmid or siRNA. As shown in
Fig. 5A and 5B, the mRNA and
protein expression levels of APLNR were increased in the EPCs
transfected with the APLNR plasmid, but reduced in the EPCs
transfected with APLNR siRNA. The proliferation of EPCs cultured
under hypoxia or normoxia was then determined using an MTT assay.
As shown in Fig. 5C, the
proliferation of EPCs in the normoxia + APLNR group was higher than
that in the normoxia group, suggesting that upregulation of APLNR
enhanced EPC proliferation. In addition, the proliferation of EPCs
in the hypoxia + APLNR siRNA group was lower than that in the
hypoxia group, suggesting that the inhibition of APLNR suppressed
hypoxia-induced EPC proliferation. Taken together, these findings
suggested that Apelin/APLNR signaling is a key regulator of
hypoxia-induced EPC proliferation.
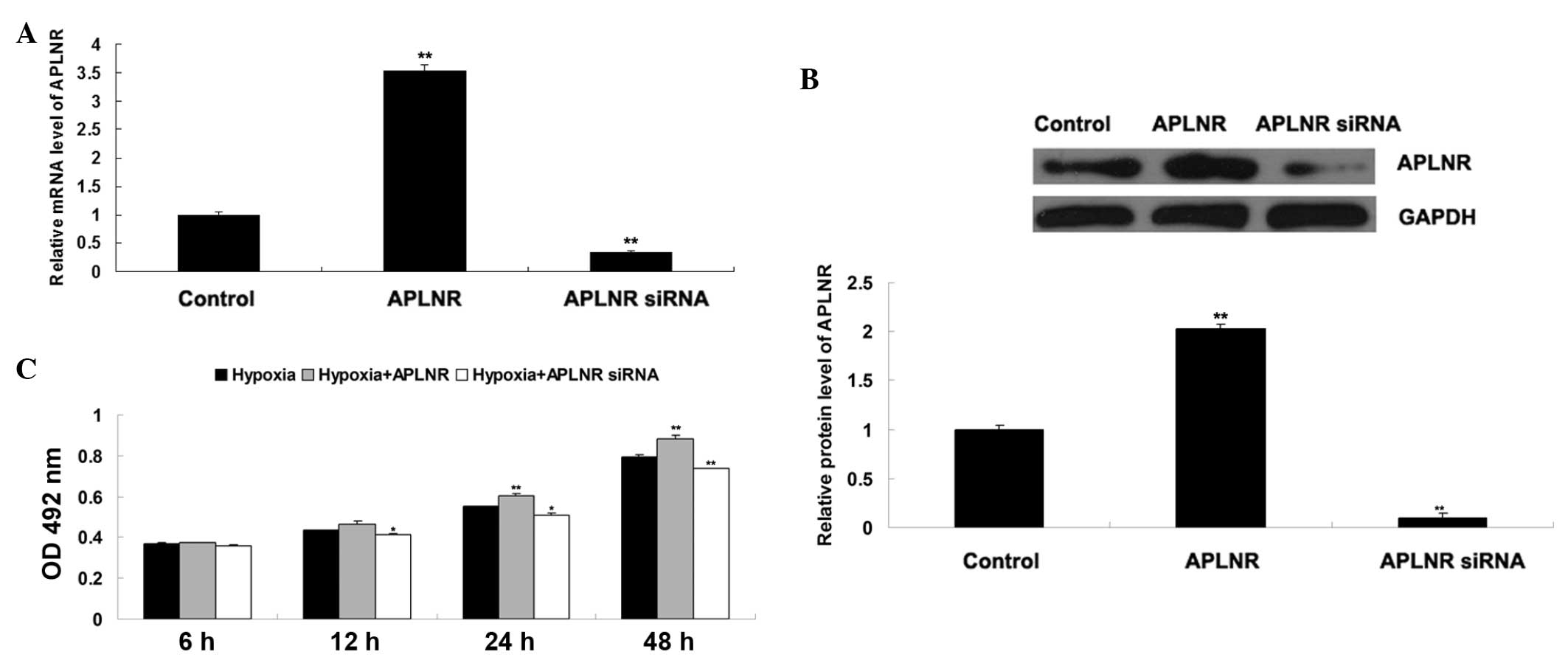 | Figure 5(A) Reverse transcription-quantitative
polymerase chain reaction was performed to examine the mRNA
expression of APLNR in EPCs transfected with either tha APLNR
plasmid or APLNR siRNA. GAPDH was used as an internal reference.
Untransfected cells were used as a control. **P<0.01
vs. control. (B) Western blotting was performed to examine the
protein expression of APLNR in EPCs transfected with either the
APLNR plasmid or APLNR siRNA, GAPDH was used as an internal
reference. Untransfected cells were used as a control.
**P<0.01, vs. control. (C) MTT assay was performed to
determine the cell proliferation of EPCs in each group at 6 h, 12
h, 24 h and 48 h. Hypoxia, EPCs cultured under hypoxia; Hypoxia +
APLNR, EPCs transfected with APLNR plasmid and cultured under
hypoxia; Hypoxia + APLNR siRNA, EPCs transfected with APLNR siRNA
and cultured under hypoxia. *P<0.05, vs. hypoxia.
**P<0.01, vs. hypoxia. Data are expressed as the mean
± standard deviation. EPC, endothelial progenitor cell; APLNR,
Apelin receptor; OD. optical density; siRNA, small interfering
RNA. |
MAPK signaling pathway is a downstream
effecter of Apelin/APLNR signaling during hypoxia-induced EPC
proliferation
As the MAPK signaling pathway has been suggested to
be involved in hypoxia-induced EPC proliferation, the present study
further investigated whether the MAPK signaling pathway is a
downstream effecter of Apelin/APLNR signaling during
hypoxia-induced EPC proliferation. The P38 MAPK inhibitor,
SB-239063, or ERK MAPK inhibitor, PD98059, was used to treat EPCs,
which had been transfected with the Apelin plasmid. A total of four
groups of EPCs were cultured under hypoxia, as follows: Control
group without any treatment, Apelin group, Apelin + SB-239063
group, and Apelin + PD98059 group. As shown in Fig. 6, the upregulation of Apelin
promoted EPC proliferation under hypoxia, which was abrogated by
pretreatment with SB-239063 or PD98059, respectively. These
findings suggested that inhibition of MAPK signaling suppressed
hypoxia-induced EPC proliferation. Taken together, it was suggested
that MAPK pathway is a downstream effecter of Apelin/APLNR
signaling during hypoxia-induced EPC proliferation.
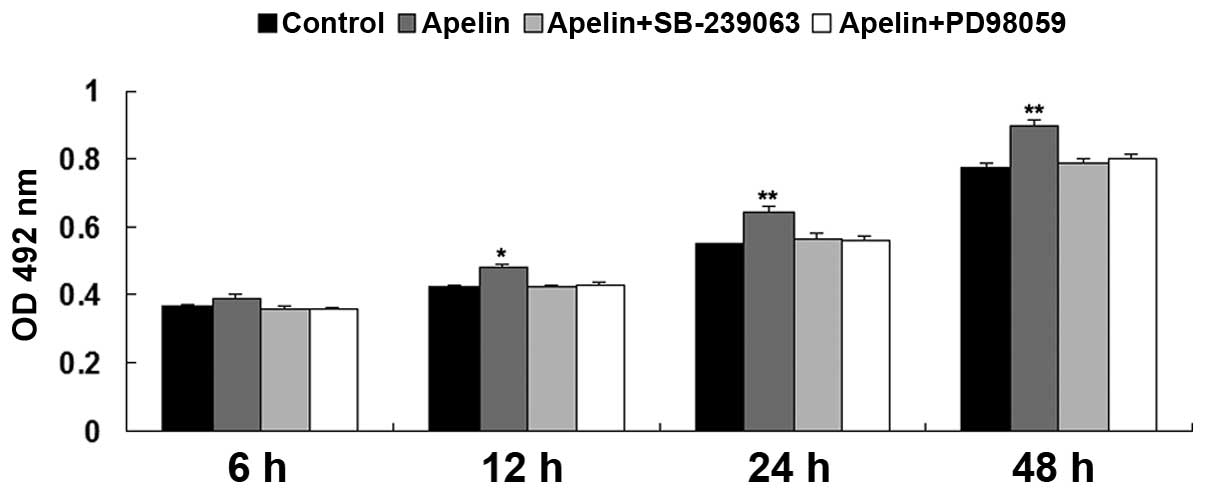 | Figure 6MTT assay was performed to determine
the cell proliferation of EPCs in each group at 6 h, 12 h, 24 h and
48 h. Control, EPCs cultured under hypoxia; Apelin, EPCs cultured
under hypoxia; Apelin + SB-239063, EPCs transfected with the Apelin
plasmid were treated with SB-239063 and cultured under hypoxia.
Apelin + PD98059, EPCs transfected with the Apelin plasmid were
treated with PD98059 and cultured under hypoxia.
*P<0.05 vs control; **P<0.01, vs.
control. Data are expressed as the mean ± standard deviation. EPC,
endothelial progenitor cell; APLNR, Apelin receptor; OD, optical
density. |
Discussion
The Apelin/APLNR signaling pathway has been
demonstrated to be closely associated with cardiovascular functions
in the systemic circulation (13).
In the present study, hypoxia-induced upregulation of HIF-α and
Apelin/APLNR signaling was found to be involved in the
proliferation of EPCs. In addition, the results suggested that MAPK
signaling is involved in Apelin/APLNR-mediated EPC
proliferation.
Hypoxia has been demonstrated to be involved in the
regulation of cellular processes of EPCs (14). In the present study, hypoxia
notably enhanced the proliferation of EPCs. Lee et al
reported that hypoxia inhibits senescence of EPCs in aged humans
(15). In addition,
hypoxia/ischemia triggers EPCs to proceed from bone marrow into the
peripheral blood, and EPCs can differentiate into ECs following
migration into the site of hypoxia/ischemia in tissues (16). In addition, HIF-1α pis important in
the regulation of angiogenesis. Overexpression of HIF-1α can
promote the differentiation of EPCs into ECs, while inhibition of
the expression of HIF-1α suppresses the differentiation of EPCs
into ECs, as well as the expression of vascular endothelial growth
factor (VEGF), VEGF receptor 2, eNOS, and the production of NO
(4,17,18).
The present study demonstrated that the expression level of HIF-α
was notably upregulated in EPCs cultured under hypoxia.
Apelin/APLNR signaling is involved in various
physiological and pathological functions in the cardiovascular
system. Apelin/APLNR signaling promotes angiogenesis and improves
cardiac functional recovery post-myocardial infarction (19). In addition, Lv et al
observed that Apelin/APLNR signaling regulates the proliferation of
vascular smooth muscle cells (20), and Kasai et al found that
inhibition of Apelin/APLNR signaling inhibits the proliferation of
ECs (21). In the present study,
the expression levels of Apelin and APLNR were found to be
increased in EPCs cultured under hypoxia. The Apelin/APLNR
signaling is also involved in the mobilization of EPCs following
acute myocardial infarction (7).
The present study hypothesized that the increased expression of
Apelin/APLNR is associated with the upregulation of HIF-α in
hypoxia-treated EPCs, as the association between HIF-1α and
Apelin/APLNR signaling has been reported in other cell types.
Glassford et al demonstrated that HIF-1α regulated the
expression of Apelin in hypoxia-treated adipocytes (22). Ronkainen et al found that
the upregulation of Apelin was eliminated by HIF inhibitory PAS
protein in cardiomyocytes cultured under hypoxia (23). These findings suggest that the
Apelin/APLNR signaling is mediated by the HIF pathway in adipocytes
and cardiomyocytes cultured under hypoxia. In addition, the present
study further demonstrated that knockdown of Apelin/APLNR signaling
significantly inhibited hypoxia-induced EPC proliferation. These
findings are the first time, to the best of our knowledge, to
suggest that Apelin/APLNR signaling is involved in regulating the
proliferation of EPCs cultured under hypoxia.
The underlying mechanisms by which Apelin/APLNR
signaling regulates hypoxia-induced EPCs proliferation requires
further investigation. The present study demonstrated that
inhibition of the p38 and ERK MAPK signaling pathways suppressed
hypoxia-induced proliferation of EPCs transfected with an Apelin
plasmid. Similar findings have been observed in other cell types.
Apelin/APLNR suppresses the production and release of reactive
oxygen species and promotes the expression of anti-oxidant enzymes
via the MAPK signaling pathway in adipocytes (24). Yang et al revealed that
Apelin/APLNR signaling protects the brain against
ischemia/reperfusion injury via activation of the ERK MAPK
signaling pathway (25). However,
whether the MAPK signaling pathway is involved in Apelin/APLNR
signaling-mediated EPC proliferation remains to be elucidated. The
data of the present study suggested, for the first time that the
p38 and ERK MAPK signaling pathway acts as a downstream effecter of
Apelin/APLNR signaling during hypoxia-induced EPC
proliferation.
In conclusion, the results of the present study
suggested that hypoxia-induced upregulation of HIF-1α activates
Apelin/APLNR signaling, which promotes the proliferation of EPCs
via the downstream MAPK signaling pathway. These findings suggested
that the Apelin/APLNR signaling may be applied as a therapeutic
target for hypoxic/ischemic injury.
References
|
1
|
Endemann DH and Schiffrin EL: Endothelial
dysfunction. J Am Soc Nephrol. 15:1983–1992. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schröder K, Kohnen A, Aicher A, Liehn EA,
Büchse T, Stein S, Weber C, Dimmeler S and Brandes RP: NADPH
oxidase Nox2 is required for hypoxia-induced mobilization of
endothelial progenitor cells. Circ Res. 105:537–544. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Simons D, Grieb G, Hristov M, Pallua N,
Weber C, Bernhagen J and Steffens G: Hypoxia-induced endothelial
secretion of macrophage migration inhibitory factor and role in
endothelial progenitor cell recruitment. J Cell Mol Med.
15:668–678. 2011. View Article : Google Scholar
|
|
4
|
Jiang M, Wang CQ, Wang BY and Huang DJ:
Inhibitory effect of siRNA targeting HIF-1alpha on differentiation
of peripheral blood endothelial progenitor cells. Ai Zheng.
24:1293–1300. 2005.In Chinese.
|
|
5
|
Kleinz MJ and Baxter GF: Apelin reduces
myocardial reperfusion injury independently of PI3K/Akt and P70S6
kinase. Regul Pept. 146:271–277. 2008. View Article : Google Scholar
|
|
6
|
Zhang Z, Yu B and Tao GZ: Apelin protects
against cardiomyocyte apoptosis induced by glucose deprivation.
Chin Med J (Engl). 122:2360–2365. 2009.
|
|
7
|
Ye J, Ni P, Kang L and Xu B: Apelin and
vascular endothelial growth factor are associated with mobilization
of endothelial progenitor cells after acute myocardial infarction.
J Biomed Res. 26:400–409. 2012. View Article : Google Scholar
|
|
8
|
Wang W, McKinnie SM, Patel VB, Haddad G,
Wang Z, Zhabyeyev P, Das SK, Basu R, McLean B, Kandalam V, et al:
Loss of Apelin exacerbates myocardial infarction adverse remodeling
and ischemia-reperfusion injury: Therapeutic potential of synthetic
Apelin analogues. J Am Heart Assoc. 2:e0002492013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kyriakis JM and Avruch J: Mammalian MAPK
signal transduction pathways activated by stress and inflammation:
A 10-year update. Physiol Rev. 92:689–737. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qiu C, Xie Q, Zhang D, Chen Q, Hu J and Xu
L: GM-CSF induces cyclin D1 expression and proliferation of
endothelial progenitor cells via PI3K and MAPK signaling. Cell
Physiol Biochem. 33:784–795. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang J, Liu Q, Hu X, Fang Z, Huang F,
Tang L and Zhou S: Apelin/APJ signaling promotes hypoxia-induced
proliferation of endothelial progenitor cells via
phosphoinositide-3 kinase/Akt signaling. Mol Med Rep. 12:3829–3834.
2015.PubMed/NCBI
|
|
12
|
Zhong Y, Lin Y, Shen S, Zhou Y, Mao F,
Guan J and Sun Q: Expression of ALDH1 in breast invasive ductal
carcinoma: An independent predictor of early tumor relapse. Cancer
Cell Int. 13:602013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
O'Carroll AM, Lolait SJ, Harris LE and
Pope GR: The Apelin receptor APJ: Journey from an orphan to a
multifaceted regulator of homeostasis. J Endocrinol. 219:R13–R35.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dai T, Zheng H and Fu GS: Hypoxia confers
protection against apoptosis via the PI3K/Akt pathway in
endothelial progenitor cells. Acta Pharmacol Sin. 29:1425–1431.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee SH, Lee JH, Yoo SY, Hur J, Kim HS and
Kwon SM: Hypoxia inhibits cellular senescence to restore the
therapeutic potential of old human endothelial progenitor cells via
the hypoxia-inducible factor-1alpha-TWIST-p21 axis. Arterioscler
Thromb Vasc Biol. 33:2407–2414. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Machalińska A: The role of circulating
endothelial progenitor cells in the development of vascular retinal
diseases. Klin Oczna. 115:158–162. 2013.In Polish.
|
|
17
|
Jiang M, Wang B, Wang C, He B, Fan H, Guo
TB, Shao Q, Gao L and Liu Y: Inhibition of hypoxia-inducible
factor-1alpha and endothelial progenitor cell differentiation by
adenoviral transfer of small interfering RNA in vitro. J Vasc Res.
43:511–521. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jiang M, Wang CQ, Wang BY, He B, Shao Q
and Huang DJ: Overexpression of hypoxia inducible factor-1alpha
(HIF-1alpha) promotes the differentiation of endothelial progenitor
cell ex vivo. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 14:565–570.
2006.In Chinese. PubMed/NCBI
|
|
19
|
Li L, Zeng H and Chen JX: Apelin-13
increases myocardial progenitor cells and improves repair
postmyocardial infarction. Am J Physiol Heart Circ Physiol.
303:H605–H618. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lv D, Li H and Chen L: Apelin and APJ, a
novel critical factor and therapeutic target for atherosclerosis.
Acta Biochim Biophys Sin (Shanghai). 45:527–533. 2013. View Article : Google Scholar
|
|
21
|
Kasai A, Ishimaru Y, Higashino K,
Kobayashi K, Yamamuro A, Yoshioka Y and Maeda S: Inhibition of
Apelin expression switches endothelial cells from proliferative to
mature state in pathological retinal angiogenesis. Angiogenesis.
16:723–734. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Glassford AJ, Yue P, Sheikh AY, Chun HJ,
Zarafshar S, Chan DA, Reaven GM, Quertermous T and Tsao PS: HIF-1
regulates hypoxia- and insulin-induced expression of Apelin in
adipocytes. Am J Physiol Endocrinol Metab. 293:E1590–E1596. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ronkainen VP, Ronkainen JJ, Hänninen SL,
Leskinen H, Ruas JL, Pereira T, Poellinger L, Vuolteenaho O and
Tavi P: Hypoxia inducible factor regulates the cardiac expression
and secretion of Apelin. FASEB J. 21:1821–1830. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Than A, Zhang X, Leow MK, Poh CL, Chong SK
and Chen P: Apelin attenuates oxidative stress in human adipocytes.
J Biol Chem. 289:3763–3774. 2014. View Article : Google Scholar :
|
|
25
|
Yang Y, Zhang X, Cui H, Zhang C, Zhu C and
Li L: Apelin-13 protects the brain against ischemia/reperfusion
injury through activating PI3K/Akt and ERK1/2 signaling pathways.
Neurosci Lett. 568:44–49. 2014. View Article : Google Scholar : PubMed/NCBI
|