Introduction
Osteosarcoma (OSA) is globally one of the most
common types of primary bone tumor and is predominantly observed in
children and adolescents (<20 years old) (1). Patients with localized disease have a
five-year recurrence-free survival rate of 80%; however, the
prognosis of OSA is poor in metastatic osteosarcoma. In spite of
OSA occurring in any type of bone in the body, the metaphyseal
(actively growing) regions of the distal femur, proximal tibia and
proximal humerus are the most frequent origins of the primary tumor
and the sites with the highest probability of metastasis are the
lungs and distant bones (2).
It has been reported that several genes are able to
regulate cell proliferation and differentiation; these genes carry
numerous mutations associated with significant neoplasmic
abnormalities in OSA (3–9). Of note, mutations in tumor suppressor
genes, including p53, MDM2 and riboblastoma protein have been
reported to have major roles in the tumorigenesis of OSA (3,4). OSA
is also associated with the aberrant expression of certain
transcription factors expressed in bones, including c-fos, whose
overexpression has been shown to result in OSA in the bones of mice
(5), as well as osteoblast
differentiation factor osterix (6,7). In
OSA cell lines, Runx2 was found to be downregulated or
dysfunctional (8), and in
high-grade pediatric OSA, genomic aberrations in the Twist have
been reported (9).
Resistance to conventional chemotherapy is one of
the characteristics of metastatic OSA and represents a considerable
obstacle for its clinical treatment (10). However, only a small number of
genes, including HES1 (11–13)
and Ezrin (10) have been
implicated in the progression and metastasis of OSA.
It has been reported that statins exert anti-tumoral
effects on OSA cells (13–15). Cystein-rich protein 61 (Cyr61), a
member of the Cyr61/connective tissue growth factor
(CTGF)/nephroblastoma overexpressed (NOV) (CCN) family of secreted
proteins, was among the factors downregulated by statins. This CCN
protein family comprises Cyr61, CTGF, NOV and Wnt-induced secreted
proteins (WISP)1, -2 and -3 (16).
As a member of the CCN family, CTGF was hypothesized have effects
on osteocarcinoma similar to those of statins. The present study
therefore assessed the effects of CTGF knockdown or
lentivirus-mediated overexpression of CTGF as well as statin
treatment on the biological properties of OSA cells.
Materials and methods
Cell lines and culture
The SaOS2, U2OS, MG63, OHS4 and CAL72 cell lines
(American Type Culture Collection, Manassas, VA, USA) were cultured
in Dulbecco's modified Eagle's medium (Invitrogen; Thermo Fisher
Scientific, Waltham, MA, USA) supplemented with 10% fetal calf
serum (FCS; Thermo Fisher Scientific) at 37°C in a humidified
atmosphere containing 5% CO2 in air.
RNA extraction and reverse-transcription
quantitative polymerase chain reaction (RT-qPCR)
TRIzol reagent (Invitrogen; Thermo Fisher
Scientific) was used to isolate RNA according to the manufacturer's
instructions, which was stored at −20°C. cDNA was synthesized using
3 µg RNA, which was denaturated and reverse-transcribed by
using 300 U Moloney murine leukemia virus reverse transcriptase, 15
mg oligo dT primers and 1 mM deoxynucleoside triphosphate (dNTP)
(Promega, Madison, WI, USA) in a total volume of 30 µl. SYBR
Green Master Mix kit (ABGen, Courtaboeuf, France) was used for
qPCR. A total of 0.5 mM of each primer (Invitrogen; Thermo Fisher
Scientific) was used with sequences as follows: Human CTGF, forward
5′-CAG GCT AGA GAA GCA GAG CC-3′ and reverse 5′-GTA ATG GCA GGC ACA
GGT CT-3′; β-actin, forward 5′-CTC CAT CCT GGC CTC GCT GT-3′ and
reverse 5′-GCT GTC ACC TTC ACC GTT CC-3′. Thermocycling was
conducted using the ABI 7500 (Applied Biosystems; Thermo Fisher
Scientific) and the cycling conditions were as follows: Initial
denaturation at 95°C for 15 min, followed by 40 cycles of 20 sec at
95°C, 15 sec at 58°C and 15 sec at 72°C, and final extension at
72°C for 7 min. The 2−ΔΔCt method was used to determine
the relative quantities of RNA.
Plasmid transduction
In order to investigate the role of CTGF in OSA,
cell lines were transduced with lentiviral vectors (LV) encoding
either the full-length sequence (LV-CTGF) or a specific short
hairpin (sh)RNA (sh-CTGF). The full-length CTGF ORF (1047 base
pairs; GenBank accession number, CR541759.1) was amplified from the
pFLAG-CMV2-CTGF plasmid (Invitrogen; Thermo Fisher Scientific). The
primer sequences were as follows: Forward,
5′-TACTGGCGGCGGTATACCCG-3′ and reverse, 5′-TGCCATGTCTCCGTACAT-3′.
The PCR product was inserted into the expression vector
pcDNA3.1/myc-His(-)B-3X FLAG-IRES-hrGFP, derived from
pcDNATM3.1/myc-His(-)B (Invitrogen; Thermo Fisher Scientific). Cell
transduction was performed using Lipofectamine 2000 (Invitrogen;
Thermo Fisher Scientific) according to the manufacturer's
instructions.
Proliferation assay
A bromodeoxyuridine (BrdU) incorporation assay was
used to quantify cell replication. A previously described procedure
was used in the present study (17). In brief, cells were cultured for 24
h in the presence of increasing concentrations of bisphosphonates
(10−9–10−4 M) and labeled with BrdU for the
last 6 h (kit purchased from GE Healthcare Life Sciences,
Roosendaal, The Netherlands).
Detection of apoptosis and necrosis
Double staining with ethidium bromide and acridine
orange was performed to visualize and quantify the number of viable
cells (green nuclei), apoptotic cells (nuclei condensed and colored
orange), and necrotic cells (red nuclei). In briefly, 2 µl
dye mixture (100 µg/ml acridine orange and 100 µg/ml
ethidium bromide) was added to 20 µl cell suspension and
immediately examined with the 40X oil immersion objective using a
Leitz DMRB fluorescence microscope (green/red filter; 100 W lamp;
Leica Microsystems GmbH, Wetzlar, Germany) equipped with a
photometrics CCD camera and the Logikon image analysis system
(Numeris Benelux SA, Ath, Belgium). Several fields, randomly
chosen, were digitized and 600–800 nuclei for each sample were
counted and scored. Results were expressed as the relative
percentages of viable, apoptotic and necrotic cells to the total
number of cells scored.
Caspase activity
Effector caspase activity was performed as
previously described (14,15). In brief, cells were treated with 10
mM atorvastatin (Adooq BioScience LLC, Irvine, CA, USA) or the
solvent for 24 h then the caspase activity was determined. Cellular
extracts (50 µg) were incubated with 0.2 mM
acetyl-Asp-Glu-Val-Asp-p-nitroanilide (caspases-3, -6 and -7; Enzo
Life Sciences, Inc., Farmingdale, NY, USA), Ac-LEHD-pNA (caspase-9;
Enzo Life Sciences, Inc.) or Ac-IETD-pNA (caspase-8; Enzo Life
Sciences, Inc.) as the substrates for the previously reported times
(14,15) at 37°C in the presence or the
absence of the specific caspase inhibitors Ac-DEVD-CHO, Ac-LEHD-CHO
and Ac-IETD-CHO (10 µM). The specific activity (nmol of
pNA/min/mg protein) was expressed as treated over control
ratios.
Migration and invasion assays
A wound-healing assay was performed following the
manufacturer's instructions (ibidi GmbH, Martinsried, Germany). A
Transwell migration and invasion assay as performed as described
previously (14). In brief, the
cells (50,000 cells/insert) were incubated 2 h with or without
statin and/or z-VAD-fmk prior to seeding into the inserts and
incubation for a further 22 h. The cells that did not migrate
through the filter were removed from the upper surface of the
membrane using cotton-tipped swabs. The cells migrated to the
underside were fixed in 3.7% paraformaldehyde in phosphate-buffered
saline (PBS) at 4°C and stained with crystal violet (Amresco,
Solon, OH, USA). The membranes were then cut from the insert and
observed under a microscope (Axioplan 2 Imaging Mot Microscope
System; Zeiss, Oberkochen, Germany). Five fields were randomly
selected and counted and each assay was performed in duplicate.
Western blot analysis
A protocol of a previous study was used for the
preparation of cell lysates (14)
In brief, the proteins (30 µg) were resolved using 12%
sodium dodecyl sulfate polyacrylamide gel electrophoresis
(SDS-PAGE; Protogel, Atlanta, GA, USA) and transferred onto
polyvinylidene difluoride nitrocellulose membranes (EMD Millipore,
Billerica, MA, USA). The filters were incubated at room temperature
for 2 h in 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 0.1% (v/v) Tween
20, 0.5% (w/v) bovine serum albumin (TBST/BSA) and then overnight
at 4°C on a shaker with the rabbit monoclonal anti-GAPDH (ab181602)
and anti-CTGF (ab6992) antibodies (1:1,000 in TBST/BSA; Abcam,
Cambridge, UK). The membranes were washed twice with TBST and
incubated for 2 h with the horseradish peroxidase-conjugated
secondary antibody (1:20,000 in TBST/BSA). Following the final
washes, the signals were visualized with Enhanced Chemiluminescence
Western Blotting Detection Reagent (GE Healthcare Life Sciences)
and autoradiographic film (X-Omat AR; Kodak, Rochester, NY, USA).
Densitometric analysis using ImageQuant software was performed
following digital scanning (Odyssey® Fc; Agfa-Gevaert,
Mortsel, Belgium).
Immunoblot analysis
A protocol of a previous study was used for the
preparation of cell lysates (14,15).
Incubation with rabbit monoclonal anti-GAPDH (ab181602; 1:200) and
rabbit polyclonal anti-CTGF (ab6992; 1:200) antibodies was
conducted at 4°C overnight. Cell extracts were collected in 2X
loading lysis buffer [50 mM Tris-HCl (pH 6.8), 2% SDS, 10%
2-mercaptoethanol, 10% glycerol and protease inhibitor cocktail;
Sigma-Aldrich, St. Louis, MO, USA]. The total cellular proteins
were separated using 8% SDS–PAGE and transferred to Hybond-C
nitrocellulose membranes (GE Healthcare Life Sciences, Chalfont,
UK). Subsequent to blocking with PBS containing 5% BSA or nonfat
milk, the membranes were incubated with the primary antibodies,
followed by incubation with IRDye 800CW or 680RD secondary
antibodies (1:10,000; LI-COR Biosciences, Lincoln, NE, USA). The
protein bands were detected using the Odyssey Infrared Imaging
System (Li-COR Biosciences).
Statistical analysis
Values are expressed as the mean ± standard
deviation. Two-factor analysis of variance was used to compare
values between groups, using SPSS software, version 19.0 (IBM SPSS,
Armonk, NY, USA). P<0.05 was considered to indicate a
statistically significant difference between values.
Results
CTGF expression is reduced by
atorvastatin (statin) in OSA cells
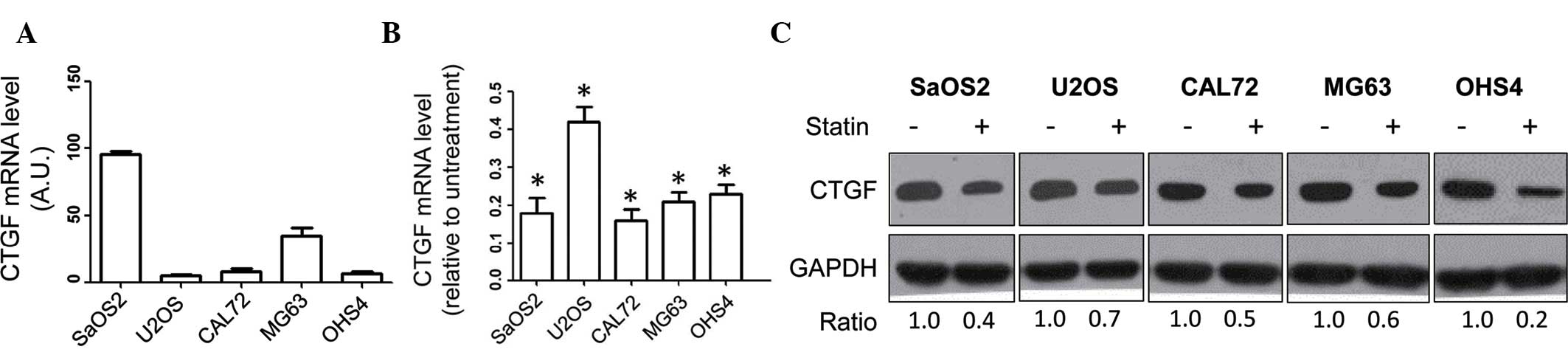
RT-qPCR analysis of CTGF was performed in the SaOS2,
U2OS, CAL72, MG63 and OHS4 human OSA cell lines, revealing that
CTGF mRNA was expressed in all cell lines, particularly in SaOS2
cells (Fig. 1A). Furthermore, the
effect of statin treatment on the expression of CTGF was assessed
in the OSA cell lines. CTGF mRNA expression in the panel of OSA
cell lines was markedly decreased following treatment with statin
(10 mM) (P<0.05 vs. untreated) (Fig. 1B). In addition, the effect of
statin (10 mM) on the protein levels of CTGF in the panel of cell
lines was assessed by immunoblot analysis, revealing that the
protein levels of CTGF were decreased following statin (Fig. 1C). Collectively, these results
indicated that statin treatment led to the downregulation of CTGF
in human OSA cells. As the SaOS2 and U2OS cell lines expressed the
highest and lowest levels of CTGF, respectively, they were selected
to be used in the subsequent experiments.
CTGF expression does not affect OSA cell
proliferation
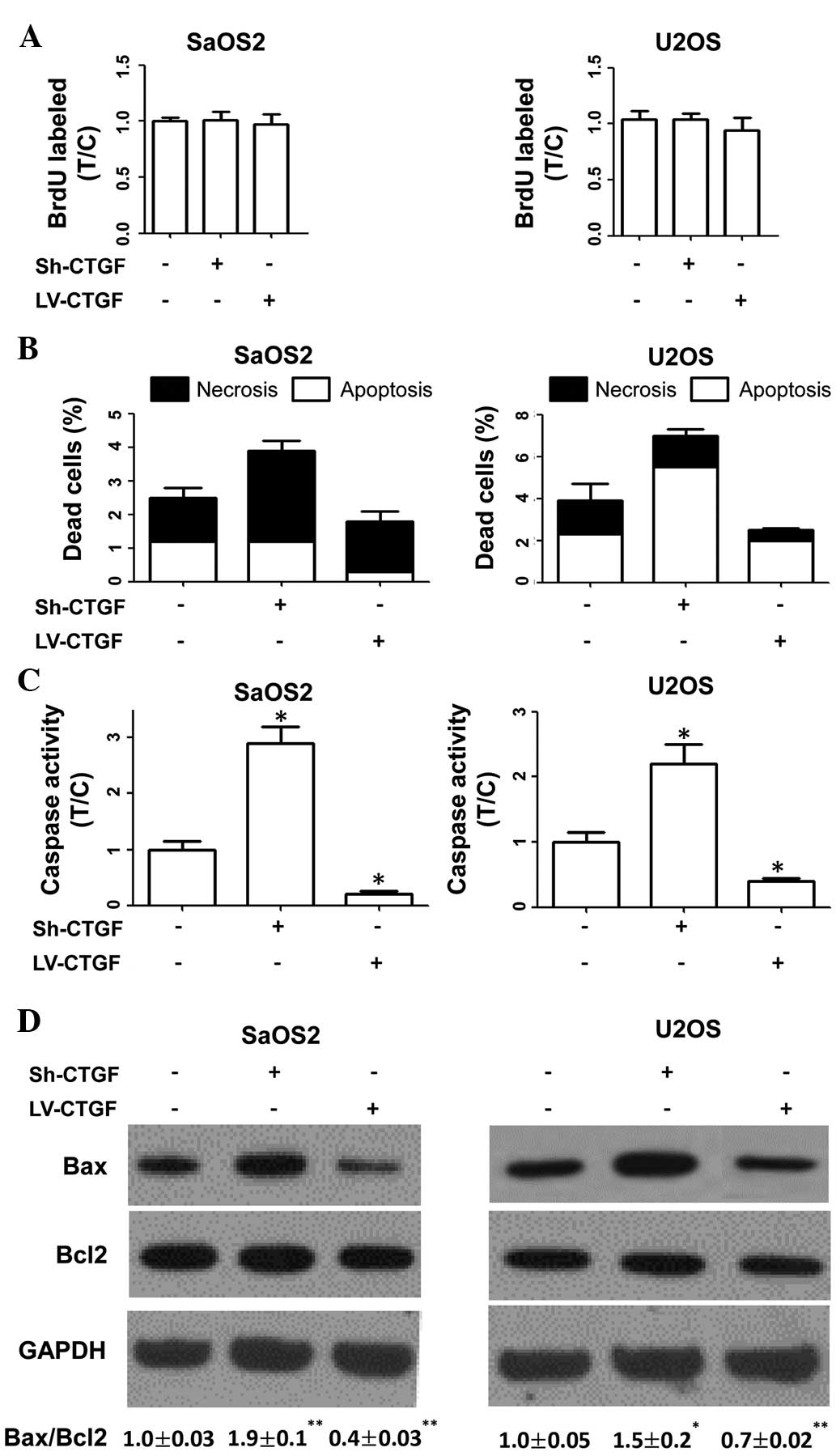
A BrdU incorporation assay were used to determine
the proliferative rates of transduced and parental cells, revealing
that these were not affected by plasmid transduction (Fig. 2A). The results therefore indicated
that CTGF had no significant effects on OSA-cell proliferation in
human cell lines.
Evasion of apoptosis by OSA cells is
dependent on CTGF expression
The present study investigated the effects of CTGF
on OSA cell death. As shown in Fig.
2B, apoptotic and necrotic indices of sh-CTGF-transduced cells
were higher than those of parental cells. By contrast,
LV-CTGF-transduced cells displayed lower apoptotic and necrotic
indices compared with those of parental cells. Furthermore, it was
revealed that sh-CTGF-transduced cells exhibited increased caspase
activity and an elevated Bax/Bcl2 ratio compared with those of
parental cells. By contrast, caspase activity and the Bax/Bcl2
ratio were reduced in CTGF-overexpressing OSA cells compared with
those in parental cells (Fig. 2C and
D). These results indicated that CTGF expression was associated
with the evasion of apoptosis by OSA cells.
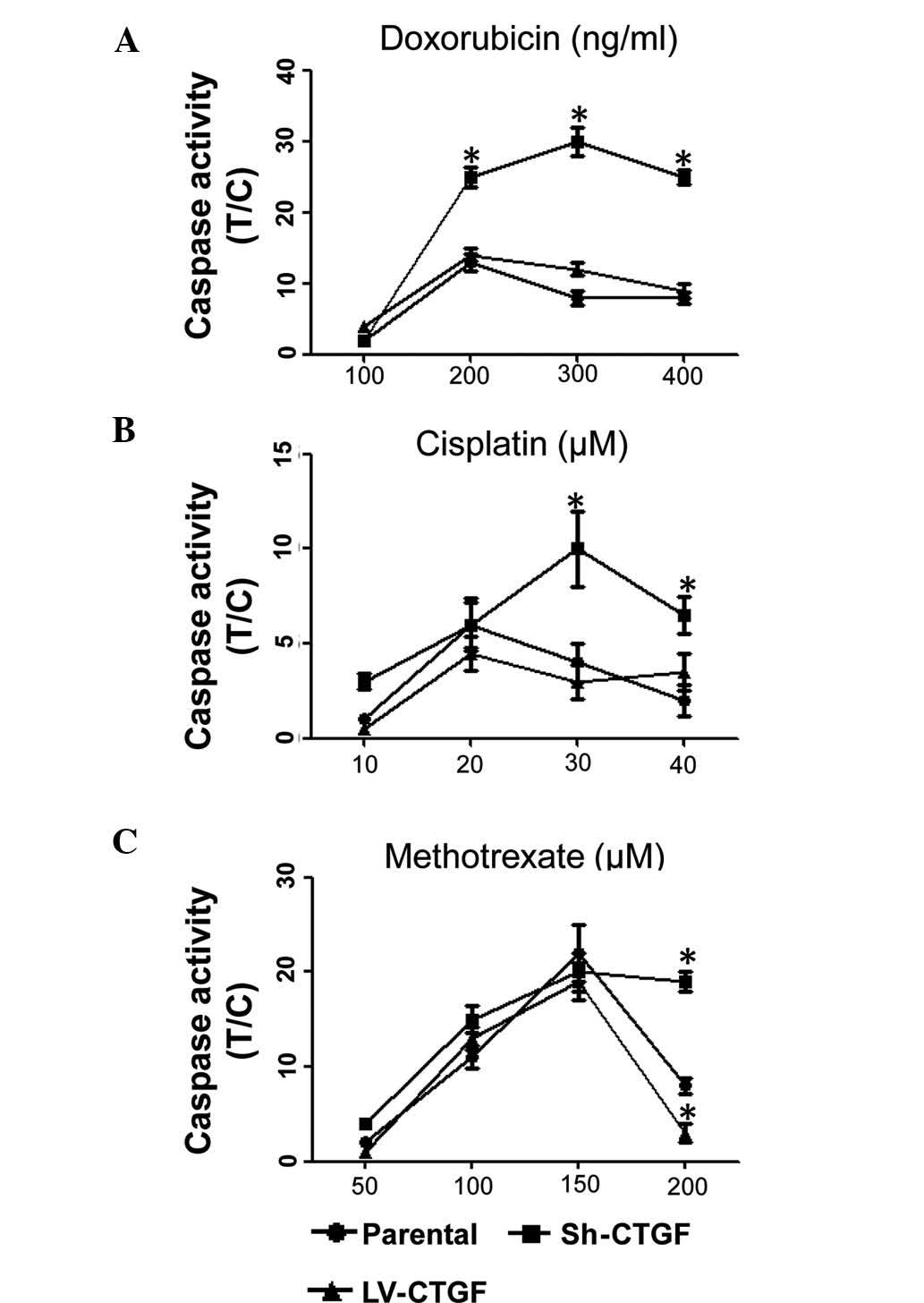
The dose-dependent cytotoxic effects of doxorubicin,
cisplatin and methotrexate on OSA cell viability are utilized for
the chemotherapeutic treatment of OSA (14). The present study revealed that CTGF
silencing significantly enhanced the caspase activity in SaOS2
cells following treatment with doxorubicin, cisplatin or
methotrexate, whereas LV-CTGF slighly decreased caspase levels
compared with those in native SaOS2 cells treated with the
chemotherapeutics (Fig. 3A–C). It
is therefore concluded that silencing of CTGF enhanced the efficacy
of chemotherapeutic drugs against OSA.
Cell migration and invasion are dependent
on CTGF expression in vitro
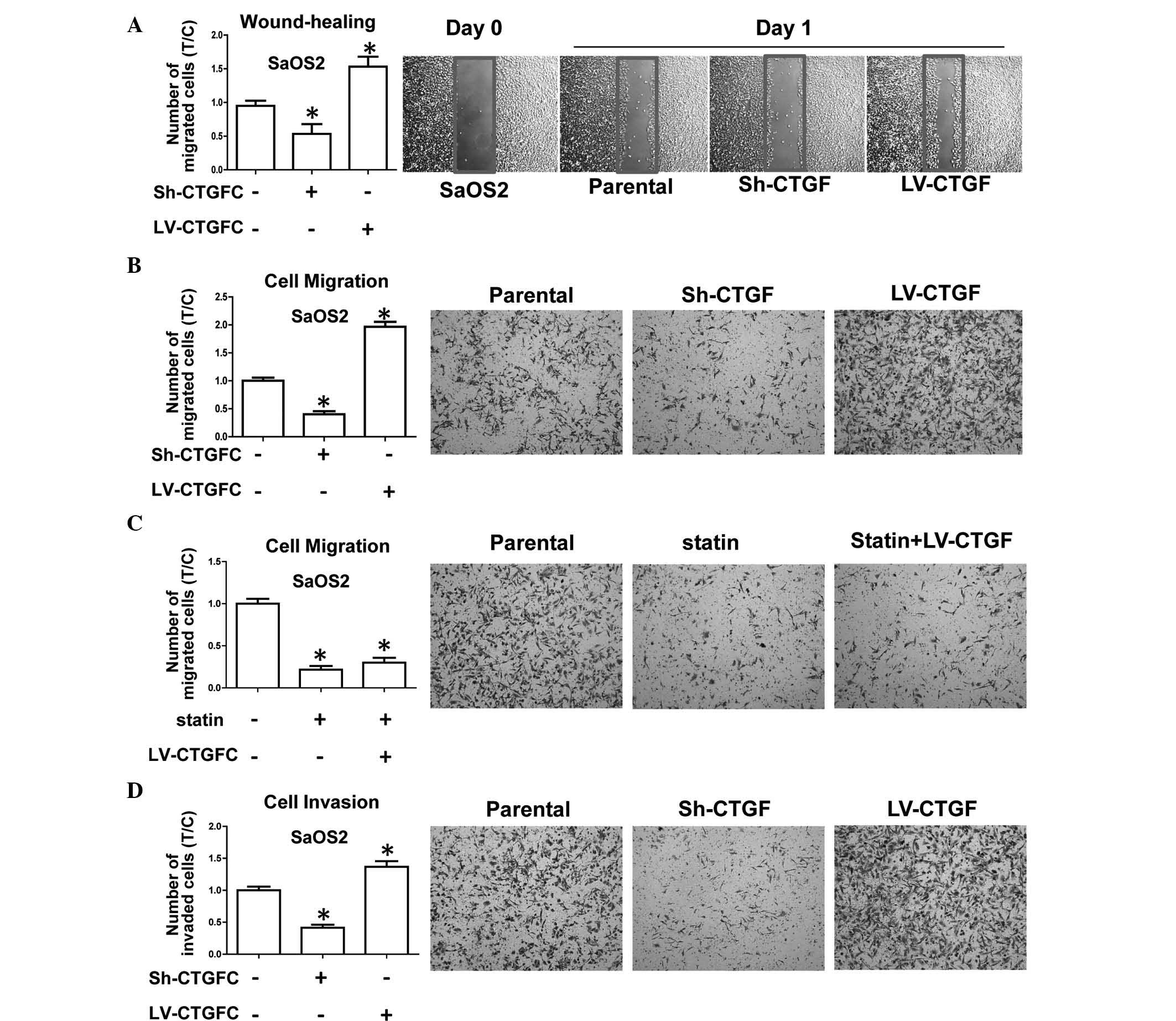
The present study further investigated the
invasiveness and migratory potential of transduced OSA cell lines,
which represent the main characteristics of OSA progression and the
development of metastasis. The results showed that CTGF silencing
inhibited wound healing in sh-CTGF-transduced cells compared with
that in parental cells, while CTGF overexpression enhanced wound
healing (Fig. 4A). In addition,
CTGF overexpression enhanced the migratory potential in a Transwell
assay (Fig. 4B). The observed
inhibition of the migratory potential by statin was not able to be
rescued by overexpression of CTGF (Fig. 4C). Furthermore, a Transwell assay
using Matrigel-coated inserts revealed that silencing of CTGF
inhibited the invasive capacity of OSA cells, while cell
invasiveness was promoted by CTGF overexpression (Fig. 4D). All of these results implied
that CTGF had positive effects on cell migration and invasiveness
in vitro, whereas invasion and migration were reduced in
CTGF-silenced OSA cells. It can be concluded that CTGF expression
is associated with the aggressiveness and metastatic potential of
OSA cells.
Discussion
Conserved cysteine residues covalently bound to
isoprenoids can be post-translationlly modified by prenylation,
which is essential for the pro-tumorigenic activity of certain
guanosine triphosphatases, including Ras and Rho-like proteins
(18,19). Synthetic bisphosphonates with
inhibitory activities on geranylgeranyltransferase type and
farnesyltransferase can be utilized as anti-cancer drugs which
partly block prenylation through inhibition of farnesyl
pyrophosphate (FPP) synthase activity; this approach is a novel
therapeutic strategy for several cancer types, including OSA and
bone metastases (20–25). Statins act as hypocholesterolemic
agents with inhibitory effects on the activity of
3-hydroxy-3-methylglutaryl-coenzyme A reductase (26) and represent another class of drug
which acts through depleting downstream isoprenoid residues,
including such as geranylgeranylpyrophosphate or FPP. Previous
studies on OSA reported that statins not only induced apoptosis but
also reduced cell migration and invasion, and potentiated the
effects of chemotherapeutic agents (13–15).
However, the anti-cancer efficacy of statins in vivo remains
to be clarified. Previous clinical studies indicated that statins,
apart from exhibiting anti-cancer effects, may also be associated
with an increased risk for the development of cancer de novo
(27–29). These conflicting results indicate
that the understanding of the mechanisms of action of statins is
required to be expanded and refined, and that novel targets for
cancer therapy require to be discovered.
Previous studies reported that Cyr61, which encodes
a secreted protein known to modulate tumor development and
progression, was downregulated by statins (30–32)
and that CTGF is also among the molecular targets of statins
(33,34). CTGF is a matricellular protein of
the CCN family of extracellular matrix-associated heparin-binding
proteins, which comprises Cyr61, CTGF, NOV and WISP1-3 (35–37).
CTGF has important roles in numerous biological processes,
including cell adhesion, migration, proliferation, angiogenesis,
skeletal development and tissue wound repair, and is critically
involved in fibrotic disease and several types of cancer (33,34,38).
Members of the CCN protein family have similar domains, indicating
that CTGF may have the similar roles in OSA cells to those of
Cyr61.
The present study enhanced or silenced the
expression of CTGF in human OSA cells to determine the role of CTGF
in OSA development and progression. A BrdU incorporation assay did
not reveal any significant effects of CTGF on the proliferation of
human OSA cell lines. By contrast, CTGF silencing slightly
increased OSA cell death and enhanced the anti-neoplasic and
pro-apoptotic effects of the chemotherapeutics doxorubicin,
cisplatin and methotrexate, which may represent a novel strategy to
enhance the efficacy of OSA treatments. A positive combinatory
effect of statins with chemotherapeutic drugs in OSA or other
cancer types has been indicated by previous studies (13,39–42).
The present study focused on CTGF expression in OSA cells,
independent of the presence of statins. As silencing of CTGF
enhanced the anti-tumoral effects chemotherapeutic drugs, it was
indicated that CTGF knockdown may reduce the resistance of OSA
cells to chemotherapy.
OSA bears the characteristics of rapid and frequent
development of metastatic lesions. In vitro experiments
performed in the present study demonstrated that the migratory and
invasive capacities of human OSA cells were reduced by CTGF
silencing, whereas CTGF overexpression led to an increase in cell
migration and invasion. By contrast, previous studies reported that
silencing or inhibition of CTGF reduced the motility and
invasiveness of breast and prostate cancer cells (43,44).
Due to this discrepancy, the roles of CCN family proteins,
particularly CTGF, in OSA require further study. In OSA cell lines,
Nov was reported to be expressed at variable levels (45) and may be associated with poor
prognosis and an increased risk of developing metastases (46). The predictive value of CTGF
expression levels with regard to the outcome and progression of
human OSA requires to be investigated in future studies analyzing
CTGF expression in primary and metastatic tumors.
In conclusion, the results of the present study
revealed that OSA cell invasion and migration was regulated by CTGF
in vitro. CTGF was indicated to have a critical role in the
genesis and progression of human OSA, and to be involved in the
evasion of apoptosis, aggressiveness and metastasis formation of
OSA. Targeting of CTGF may be a strategy to enhance the efficacy of
chemotherapeutics in the treatment of OSA as well as to reduce the
aggressiveness of OSA cells.
Abbreviations:
|
Cyr61
|
cysteine-rich angiogenic inducer
61
|
|
CTGF
|
connective tissue growth factor
|
|
NOV
|
nephroblastoma-overexpressed gene
protein homolog
|
|
OSA
|
osteosarcoma
|
|
MDM2
|
mouse double minute 2 homolog
|
|
Rb
|
retinoblastoma gene
|
|
HES1
|
hes family BHLH transcription factor
1
|
|
MMLV
|
moloney murine leukemia virus reverse
transcriptase
|
|
dNTP
|
deoxynucleoside triphosphate
|
|
qPCR
|
quantitative polymerase chain
reaction
|
|
GTPase
|
hydrolase enzymes of guanosine
triphosphate
|
|
FTase
|
farnesyltransferase
|
|
FPP
|
farnesyl pyrophosphate
|
|
HMGCoA
|
3-hydroxy-3-methyl glutaryl coenzyme
A
|
|
GGPP
|
geranylgeranyl pyrophosphate
|
References
|
1
|
Fromigue O, Hamidouche Z, Vaudin P,
Lecanda F, Patino A, Barbry P, Mari B and Marie PJ: CYR61
downregulation reduces osteosarcoma cell invasion, migration, and
metastasis. J Bone Miner Res. 26:1533–1542. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang Y, Zhang L, Zhang G, Li S, Duan J,
Cheng J, Ding G, Zhou C, Zhang J, Luo P, et al: Osteosarcoma
metastasis: Prospective role of ezrin. Tumour Biol. 35:5055–5059.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pompetti F, Rizzo P, Simon RM, Freidlin B,
Mew DJ, Pass HI, Picci P, Levine AS and Carbone M: Oncogene
alterations in primary, recurrent and metastatic human bone tumors.
J Cell Biochem. 63:37–50. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ladanyi M, Cha C, Lewis R, Jhanwar SC,
Huvos AG and Healey JH: MDM2 gene amplification in metastatic
osteosarcoma. Cancer Res. 53:16–18. 1993.PubMed/NCBI
|
|
5
|
Wang ZQ, Liang J, Schellander K, Wagner EF
and Grigoriadis AE: c-fos-induced osteosarcoma formation in
transgenic mice: Cooperativity with c-jun and the role of
endogenous c-fos. Cancer Res. 55:6244–6251. 1995.PubMed/NCBI
|
|
6
|
Cao Y, Jia SF, Chakravarty G, de
Crombrugghe B and Kleinerman ES: The osterix transcription factor
down-regulates interleukin-1 alpha expression in mouse osteosarcoma
cells. Mol Cancer Res. 6:119–126. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cao Y, Zhou Z, de Crombrugghe B, Nakashima
K, Guan H, Duan X, Jia SF and Kleinerman ES: Osterix, a
transcription factor for osteoblast differentiation, mediates
antitumor activity in murine osteosarcoma. Cancer Res.
65:1124–1128. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thomas DM, Johnson SA, Sims NA, Trivett
MK, Slavin JL, Rubin BP, Waring P, McArthur GA, Walkley CR,
Holloway AJ, et al: Terminal osteoblast differentiation, mediated
by runx2 and p27KIP1, is disrupted in osteosarcoma. J Cell Biol.
167:925–934. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Entz-Werlé N, Stoetzel C, Berard-Marec P,
Kalifa C, Brugiere L, Pacquement H, Schmitt C, Tabone MD, Gentet
JC, Quillet R, et al: Frequent genomic abnormalities at TWIST in
human pediatric osteosarcomas. Int J Cancer. 117:349–355. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
O'Farrill JS and Gordon N: Autophagy in
osteosarcoma. Adv Exp Med Biol. 804:147–160. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang P, Yang Y, Zweidler-McKay PA and
Hughes DP: Critical role of notch signaling in osteosarcoma
invasion and metastasis. Clin Cancer Res. 14:2962–2969. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Khanna C, Wan X, Bose S, Cassaday R, Olomu
O, Mendoza A, Yeung C, Gorlick R, Hewitt SM and Helman LJ: The
membrane-cytoskeleton linker ezrin is necessary for osteosarcoma
metastasis. Nat Med. 10:182–186. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fromigué O, Hamidouche Z and Marie PJ:
Statin-induced inhibition of 3-hydroxy-3-methyl glutaryl coenzyme a
reductase sensitizes human osteosarcoma cells to anticancer drugs.
J Pharmacol Exp Ther. 325:595–600. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fromigué O, Hamidouche Z and Marie PJ:
Blockade of the RhoA-JNK-c-Jun-MMP2 cascade by atorvastatin reduces
osteosarcoma cell invasion. J Biol Chem. 283:30549–30556. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fromigué O, Haÿ E, Modrowski D, Bouvet S,
Jacquel A, Auberger P and Marie PJ: RhoA GTPase inactivation by
statins induces osteosarcoma cell apoptosis by inhibiting
p42/p44-MAPKs-Bcl-2 signaling independently of BMP-2 and cell
differentiation. Cell Death Differ. 13:1845–1856. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang GP and Lau LF: Cyr61, product of a
growth factor-inducible immediate early gene, is associated with
the extracellular matrix and the cell surface. Cell Growth Differ.
2:351–357. 1991.PubMed/NCBI
|
|
17
|
Fromigue O, Lagneaux L and Body JJ:
Bisphosphonates induce breast cancer cell death in vitro. J Bone
Miner Res. 15:2211–2221. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Casey PJ and Seabra MC: Protein
prenyltransferases. J Biol Chem. 271:5289–5292. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
McTaggart SJ: Isoprenylated proteins. Cell
Mol Life Sci. 63:255–267. 2006. View Article : Google Scholar
|
|
20
|
Hiraga T, Williams PJ, Mundy GR and Yoneda
T: The bisphosphonate ibandronate promotes apoptosis in MDA-MB-231
human breast cancer cells in bone metastases. Cancer Res.
61:4418–4424. 2001.PubMed/NCBI
|
|
21
|
Lee MV, Fong EM, Singer FR and Guenette
RS: Bisphosphonate treatment inhibits the growth of prostate cancer
cells. Cancer Res. 61:2602–2608. 2001.PubMed/NCBI
|
|
22
|
Ory B, Heymann MF, Kamijo A, Gouin F,
Heymann D and Redini F: Zoledronic acid suppresses lung metastases
and prolongs overall survival of osteosarcoma-bearing mice. Cancer.
104:2522–2529. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sonnemann J, Eckervogt V, Truckenbrod B,
Boos J, Winkelmann W and van Valen F: The bisphosphonate
pamidronate is a potent inhibitor of human osteosarcoma cell growth
in vitro. Anticancer Drugs. 12:459–465. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Delarue FL, Adnane J, Joshi B, Blaskovich
MA, Wang DA, Hawker J, Bizouarn F, Ohkanda J, Zhu K, Hamilton AD,
et al: Farnesyltransferase and geranylgeranyltransferase I
inhibitors upregulate RhoB expression by HDAC1 dissociation, HAT
association and histone acetylation of the RhoB promoter. Oncogene.
26:633–640. 2007. View Article : Google Scholar
|
|
25
|
Sebti SM and Hamilton AD:
Farnesyltransferase and geranylgeranyltransferase I inhibitors and
cancer therapy: Lessons from mechanism and bench-to-bedside
translational studies. Oncogene. 19:6584–6593. 2000. View Article : Google Scholar
|
|
26
|
Istvan E: Statin inhibition of HMG-CoA
reductase: A 3-dimensional view. Atheroscler Suppl. 4:3–8. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Farwell WR, Scranton RE, Lawler EV, Lew
RA, Brophy MT, Fiore LD and Gaziano JM: The association between
statins and cancer incidence in a veterans population. J Natl
Cancer Inst. 100:134–139. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Beri A, Sural N and Mahajan SB:
Non-atheroprotective effects of statins: A systematic review. Am J
Cardiovasc Drugs. 9:361–370. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gonyeau MJ and Yuen DW: A clinical review
of statins and cancer: Helpful or harmful? Pharmacotherapy.
30:177–194. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Babic AM, Kireeva ML, Kolesnikova TV and
Lau LF: CYR61, a product of a growth factor-inducible immediate
early gene, promotes angiogenesis and tumor growth. Proc Natl Acad
Sci USA. 95:6355–6360. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bleau AM, Planque N and Perbal B: CCN
proteins and cancer: two to tango. Front Biosci. 10:998–1009. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Menéndez JA, Mehmi I, Griggs DW and Lupu
R: The angiogenic factor CYR61 in breast cancer: Molecular
pathology and therapeutic perspectives. Endocr Relat Cancer.
10:141–152. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jun JI and Lau LF: Taking aim at the
extracellular matrix: CCN proteins as emerging therapeutic targets.
Nat Rev Drug Discov. 10:945–963. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hall-Glenn F and Lyons KM: Roles for CCN2
in normal physiological processes. Cell Mol Life Sci. 68:3209–3217.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen CC and Lau LF: Functions and
mechanisms of action of CCN matricellular proteins. Int J Biochem
Cell Biol. 41:771–783. 2009. View Article : Google Scholar :
|
|
36
|
Holbourn KP, Acharya KR and Perbal B: The
CCN family of proteins: Structure-function relationships. Trends
Biochem Sci. 33:461–473. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Leask A and Abraham DJ: All in the CCN
family: Essential matricellular signaling modulators emerge from
the bunker. J Cell Sci. 119:4803–4810. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kubota S and Takigawa M: The role of CCN2
in cartilage and bone development. J Cell Commun Signal. 5:209–217.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Martirosyan A, Clendening JW, Goard CA and
Penn LZ: Lovastatin induces apoptosis of ovarian cancer cells and
synergizes with doxorubicin: Potential therapeutic relevance. BMC
Cancer. 10:1032010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Schmidmaier R, Baumann P, Bumeder I,
Meinhardt G, Straka C and Emmerich B: First clinical experience
with simvastatin to overcome drug resistance in refractory multiple
myeloma. Eur J Haematol. 79:240–243. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
van der Spek E, Bloem AC, Sinnige HA and
Lokhorst HM: High dose simvastatin does not reverse resistance to
vincristine, adriamycin and dexamethasone (VAD) in myeloma.
Haematologica. 92:e130–e131. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
van der Spek E, Bloem AC, van de Donk NW,
Bogers LH, van der Griend R, Kramer MH, de Weerdt O, Wittebol S and
Lokhorst HM: Dose-finding study of high-dose simvastatin combined
with standard chemotherapy in patients with relapsed or refractory
myeloma or lymphoma. Haematologica. 91:542–545. 2006.PubMed/NCBI
|
|
43
|
Marra M, Santini D, Meo G, Vincenzi B,
Zappavigna S, Baldi A, Rosolowski M, Tonini G, Loeffler M, Lupu R,
et al: Cyr61 down-modulation potentiates the anticancer effects of
zoledronic acid in androgen-independent prostate cancer cells. Int
J Cancer. 125:2004–2013. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nguyen N, Kuliopulos A, Graham RA and
Covic L: Tumor-derived Cyr61(CCN1) promotes stromal matrix
metalloproteinase-1 production and protease-activated receptor
1-dependent migration of breast cancer cells. Cancer Res.
66:2658–2665. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Manara MC, Perbal B, Benini S, Strammiello
R, Cerisano V, Perdichizzi S, Serra M, Astolfi A, Bertoni F, Alami
J, et al: The expression of ccn3 (nov) gene in musculoskeletal
tumors. Am J Pathol. 160:849–859. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Perbal B, Zuntini M, Zambelli D, Serra M,
Sciandra M, Cantiani L, Lucarelli E, Picci P and Scotlandi K:
Prognostic value of CCN3 in osteosarcoma. Clin Cancer Res.
14:701–709. 2008. View Article : Google Scholar : PubMed/NCBI
|