Introduction
Trauma to the basal layer can lead to the
development of intrauterine scars, resulting in Asherman syndrome
(1), which is associated with
secondary infertility and miscarriage. The risk of Asherman
syndrome has increased in recent years due to intrauterine
operations becoming increasingly common. Endometrial fibrosis is
the main pathological characteristic of Asherman syndrome, which
consists of excessive deposition and reorganization of the
extracellular matrix (ECM), in place of normal endometrium
(2). At present, surgical removal
of scar tissue and ancillary treatments, including physical
barriers and hormone therapy, are recommended to treat endometrial
fibrosis. However, the recurrence of the disease remains high,
particularly in severe cases. Therefore, novel therapies that
target the cellular mechanisms underlying the development of these
pathologies are required. Epithelial and stromal cells are two main
cell populations present in the endometrium. During homeostasis, a
subset of stromal cells differentiates and incorporates stably into
the epithelial compartment (3).
Furthermore, stromal cells are critical regulators of endometrial
function via the paracrine pathway, which regulates epithelial and
leukocyte functions (4).
Therefore, post-injury stromal cell dysfunction may have a key role
in the development of endometrial fibrosis, which is considered a
failure of tissue regeneration.
Transforming growth factor (TGF)-β1 is known to
regulate various cellular responses, including ECM production, cell
proliferation, apoptosis and differentiation (5). In the TGF-β1 pathway, Smad2 and Smad3
are receptor-regulated effector proteins (R-Smads), which are
phosphorylated by the activated TGF-β type I receptor at a
C-terminal SSXS motif, resulting in R-Smad nuclear accumulation
(6). Notably, TGF-β1 is a critical
fibrotic cytokine that has a significant role in organ fibrosis and
dysfunction. In the context of fibrosis, TGF-β1 upregulates
numerous fibrogenic genes, including collagenous ECM proteins and
α-smooth muscle actin (α-SMA), via the Smad2/3 signaling pathway
(7). It has also been demonstrated
that the concentration of TGF-β1 is significantly higher in the
intrauterine fluid of patients with endothelial fibrosis, as
compared with in healthy individuals (8). However, whether the TGF-β1/Smad
pathway is involved in endometrial fibrosis remains unclear.
The microRNA (miR)-29 family shares the same seed
binding sequence, and is well-characterized by its ability to
regulate ECM proteins, including collagens, elastin and fibrillins
(9). The miR-29 family has been
shown to be reduced in various types of tissue fibrosis, and their
anti-fibrotic role has been demonstrated in the heart (10), kidney (11), liver (12), lung (13) and peritoneum (14). Long non-coding RNAs (lncRNA) have
recently been reported to regulate gene expression. Among them,
maternally expressed gene 3 (MEG3), which may be regulated by
miR-29 (15), has been shown to
induce caspase-3-dependent apoptosis in TGF-β1-treated LX-2 cells,
thus inhibiting stellate cell activation and liver fibrosis
progression (16). In our previous
study, the expression levels of TGF-β1 were increased, whereas
miR-29 was decreased in the clinical samples of patients with
Asherman syndrome (17). However,
the mechanisms underlying the interaction between miR-29 and
TGF-β1, and the functional importance of miR-29 in endometrial
fibrosis remain unexplored. Knowledge of these mechanisms is
indispensable for the development of novel strategies for the
prevention and treatment of endometrial fibrosis.
The present study aimed to investigate the
TGF-β1-induced development of endometrial fibrosis using primary
human endometrial stromal cells (ESCs). Furthermore, the preventive
and therapeutic potential of miR-29b mimics, and the associated
underlying mechanisms, were determined in the established cell
model of endometrial fibrosis.
Materials and methods
Clinical subjects and ethics
statement
Human endometrial tissue samples were obtained by
curettage from six patients who had undergone a hysterectomy. The
patients were between 30 and 45 years old, had a normal menstrual
cycle, and underwent hysterectomy for conditions other than
endometrial disease. Patients with subserous or intramural
leiomyoma, or cervical intraepithelial neoplasia, who were
estimated to be in the mid or late proliferative phase of the
menstrual cycle were selected for the present study. The present
study was approved by the Ethics Committee of Zhujiang Hospital
affiliated to Southern Medical University (Guangzhou, China).
Primary ESC isolation, purification and
culture
The protocol for stromal cell isolation was similar
to that described in a previous study (18). Full-thickness endometrial tissue
samples were collected under aseptic conditions and placed
immediately in phosphate-buffered saline (PBS; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) containing streptomycin (100
mg/ml; Gibco; Thermo Fisher Scientific, Inc.). The samples were
transported to the laboratory within 30 min. After several washes
with PBS, the tissue samples were minced into 0.5–1 mm3
pieces, and were incubated in Dulbecco's modified Eagle's
medium/Nutrient Mixture F-12 (DMEM/F12; Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 0.2% collagenase I
(Sigma-Aldrich China, Inc., Shanghai, China) for 60 min at 37°C
with agitation. Subsequently, the resulting suspension was filtered
through sterile 100- and 40-µm nylon strainers (BD
Biosciences, Shanghai, China) in turn, in order to remove
undigested material and epithelial cells. Following centrifugation
at 150 × g for 5 min at room temperature, the supernatant was
discarded and the ESCs were resuspended in DMEM/F12 supplemented
with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific,
Inc.), plated into 25 cm2 flasks, and incubated at 37°C
in an atmosphere containing 5% CO2. After 24 h, the
medium was replaced and the unattached cells were removed. The
remaining attached stromal cells were supplemented with fresh
culture medium every three days until they reached confluence. The
3rd–6th passages were used for subsequent
experiments.
Treatment with TGF-β1
ESCs were seeded in 6-well plates (or on sterile
cover glass) at a density of 2×105 cells/well in order
to investigate the function of TGF-β1 in primary ESCs. The cells
were initially incubated with TGF-β1 (1.5 or 10 ng/ml) for 48 h and
the vehicle (10 mM citric acid; Peprotech, Inc., Rocky Hill, NJ,
USA) was used as a negative control. In addition, the ESCs were
incubated with 10 ng/ml TGF-β1 for 12, 24, 48 or 72 h. Cells in the
6-well plates were harvested for reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) and western blotting, and the
cover glass of cells were used for scanning electron
microscopy.
Transfection
The cultured ESCs were seeded into 96-well plates
(3×103 cells/well) for cell proliferation analysis, into
6-well plates (2×105 cells/well) for cell cycle
distribution and apoptosis analyses, RT-qPCR and western blotting,
or into 30 mm dishes (2×105 cells/dish) for
immunofluorescence staining. For the preventive treatment, the
cells were incubated overnight to a density of 30–50%, and were
then incubated for a further 24 h in serum-free culture media, in
order to synchronize the cell cycle. Subsequently, miR-29b mimics
and a scrambled miRNA control designed by Guangzhou RiboBio Co.,
Ltd. (Guangzhou, China) were transfected into the cells using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. A
total of 6 h post-transfection, the media were refreshed and ESCs
were stimulated with TGF-β1 (10 ng/ml; Peprotech, Inc.) for 48 h
for RT-qPCR analysis, and for 72 h for western blot analysis. For
the therapeutic treatment, ESCs were treated with TGF-β1 (10 ng/ml)
for 24 h ahead of miR-29b transfection as described. The media were
refreshed and ESCs were continuously stimulated with TGF-β1 (10
ng/ml) for 24 h for RT-qPCR, and for 48 h for western blot
analysis. For the therapeutic treatment, ESCs were initially
treated with TGF-β1 (10 ng/ml) for 24 h, then transfected with
miR-29 as described. Thereafter, the medium was changed with TGF-β1
(10 ng/ml) for another 24 h.
Immunocytochemistry
Immunocytochemical staining was performed, in order
to determine the purity of the stromal cells following isolation.
The cells were cultured on cover slides in 30 mm culture dishes
(Corning China, Shanghai, China), and fixed with 4%
paraformaldehyde. Cells were permeabilized with 0.5% Triton X-100
(Shanghai Solarbio Bioscience & Technology Co., Ltd., Shanghai,
China) for 10 min, rinsed with PBS three times, incubated with 3%
H2O2 for 15 min to remove endogenous
peroxidase and incubated for 30 min with 5% BSA for antigen
blocking (Beyotime Institute of Biotechnology). Primary antibodies
targeting mouse monoclonal cytokeratin 18 (cat. no. sc-32329; 1:400
dilution; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and
mouse monoclonal vimentin (cat. no. sc-373717; 1:400 dilution;
Santa Cruz Biotechnology, Inc.) were incubated with cells for 2 h
at room temperature followed by incubation with
peroxidase-conjugated goat anti-mouse IgG secondary antibody (cat.
no. ZDR-5307; ZSGB-BIO, Beijing, China) for 40 min at room
temperature, as previously described (19). Images were captured using a Leica
DM3000 microscope (Leica Microsystems GmbH, Wetzlar, Germany).
RNA extraction and RT-qPCR
Total RNA was extracted from the cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. All of the mRNA,
lncRNA and miRNA from each sample were reverse transcribed into
cDNA using the PrimeScript RT Reagent kit (Takara Bio Inc., Otsu,
Japan). The RNA was initially treated with gDNA Eraser according to
the manufacturer's instructions. Thereafter, the RT reaction was
conducted at 37°C for 15 min, 85°C for 5 sec and 4°C prior to qPCR
analysis. qPCR was performed using the SYBR Premix Ex Taq
(Takara Bio, Inc.). The mRNA PCR primers (Invitrogen; Thermo Fisher
Scientific, Inc.) used in the present study are summarized in
Table I. For analysis of miR-29b
expression, miRNA-specific stem-loop RT primers and qPCR primers
provided in the miRNA quantification kit (Bulge-loop™ miRNA qRT-PCR
Primer Sets, one RT primer and a pair of qPCR primers for each set)
and specific for miR-29b were used and designed by Guangzhou
RiboBio Co., Ltd. For mRNA and lncRNA, qPCR was conducted at 95°C
for 15 sec followed by 40 cycles at 95°C for 5 sec and 60°C for 60
sec, and a final extension step at 72°C for 10 sec in a Roche
LightCycler480 Real-Time PCR system (Roche Diagnostics, Basel,
Switzerland). For miR-29 and U6, qPCR was conducted at 95°C for 15
sec followed by 40 cycles at 95°C for 5 sec, 57°C for 20 sec and
72°C for 10 sec. The relative levels of the RNAs of interest were
normalized with internal controls [U6 or glyceraldehyde 3-phosphate
dehydrogenase (GAPDH)], and gene expression was analyzed using the
2−ΔΔCq method (20).
 | Table IQuantitative polymerase chain reaction
primer sequences. |
Table I
Quantitative polymerase chain reaction
primer sequences.
| Name | Sequence (5′-3′) |
|---|
| COL1A1 | F:
5′-GAGGGCCAAGACGAAGACATC-3′ |
| R:
5′-CAGATCACGTCATCGCACAAC-3′ |
| α-SMA | F:
5′-GGCTCTGGGCTCTGTAAGG-3′ |
| R:
5′-CTCTTGCTCTGGGCTTCATC-3′ |
| MEG3 | F:
5′-GCTCTACTCCGTGGAAGCAC-3′ |
| R:
5′-CAAACCAGGAAGGAGACGAG-3′ |
| GAPDH | F:
5′-CGGAGTCAACGGATTTGGTCGTAT-3′ |
| R:
5′-AGCCTTCTCCATGGTGGTGAAGAC-3′ |
Western blot analysis
Cells were scraped off the plates, centrifuged at
200 × g for 5 min at 4°C and total protein was extracted using 50
µl radioimmunoprecipitation assay buffer (Wuhan Boster
Biological Technology, Ltd., Wuhan, China) supplemented with 0.5
µl protease inhibitors (Nanjing KeyGen Biotech. Co., Ltd.,
Nanjing, China) for 30 min. The lysates were collected by
centrifugation at 13,523 × g for 20 min at 4°C. Total protein
concentrations were quantified using a bicinchoninic acid protein
assay kit (Beyotime Institute of Biotechnology, Haimen, China).
Total protein from each sample (40 µg) was separated by
10–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis
and transferred to polyvinylidene difluoride membranes (EMD
Millipore, Billerica, MA, USA). The membranes were blocked with 5%
non-fat dry milk in Tris-buffered saline 1 h prior to incubation
with the following primary antibodies: Rabbit polyclonal
anti-collagen, type 1, alpha 1 (COL1A1; 1:1,000; cat. no. ab34710;
Abcam, Cambridge, MA, USA), rabbit polyclonal anti-α-SMA (1:1,000;
cat. no. ab5694; Abcam), rabbit polyclonal anti-phosphorylated
(p)-Smad2/3 (1:1,000; cat. no. sc-11769-R; Santa Cruz
Biotechnology, Inc.) and rabbit polyclonal GAPDH (1:1,000 dilution;
Santa Cruz Biotechnology, Inc.; cat. no. sc-25778) for 12 h at 4°C.
Subsequently, the blots were incubated with peroxidase-conjugated
secondary antibody (1:5,000 dilution; cat. no. ZDR-5307; ZSGB-BIO,
Beijing, China) for 1 h at room temperature, and the proteins were
visualized using Chemiluminescent Horseradish Peroxidase Substrate
(EMD Millipore), according to the manufacturer's protocol.
Chemiluminescence measurements and semi-quantitative values were
obtained using the ChemiDoc™ XRS+ Imaging system (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) and Image Gauge V3.12
(Fujifilm, Tokyo, Japan). The densitometric analyses were repeated
three times, and protein expression levels were quantified relative
to the internal control GAPDH.
Scanning electron microscopy (SEM) of
cell morphology
The cells were fixed with cold 2.5% glutaraldehyde
in 0.1 M PBS (4°C, 24 h), and were post-fixed in 1% osmium
tetroxide with 0.1% potassium ferricyanide. The samples were
dehydrated through a graded series of ethanol concentrations, and
embedded in hexamethyldisilazane, followed by air-drying. The
samples were gold-coated prior to examination under a KYKY-EM3200
scanning electron microscope (Kyky Technology Development Co.,
Ltd., Beijing, China) at an accelerating voltage of 10 keV.
Immunofluorescence staining
ESCs were fixed and permeabilized in 0.5% Triton
X-100/PBS. Following treatment with blocking buffer (BIOSS,
Beijing, China) supplemented with 5% goat serum for 30 min, the
cells were incubated with the specific primary antibodies against
p-Smad2/3 (1:100; cat. no. sc-11769-R), α-SMA (1:100; cat. no.
ab5694) and COL1A1 (1:100; cat. no. ab34710) for 1 h at room
temperature, followed by incubation with a secondary antibody (Cy3
AffiniPure Goat Anti-Rabbit Immunoglobulin G; Earthox Life
Sciences, Milbrae, CA, USA; cat. no. E031640-01) for 30 min in the
dark. Finally, the cells were incubated with
4′,6-diamidino-2-phenylindole (BestBio, Shanghai, China) for 15
min. Images were captured under an inverted fluorescence microscope
(IX71; Olympus Corporation, Tokyo, Japan).
Cell proliferation assay
Cell counting kit-8 (CCK8) assay was performed to
analyze cell proliferation. CCK-8 solution (10 µl; Dojindo
Molecular Technologies, Inc., Kumamoto, Japan) was added to each
well, and the cells were incubated for 2 h. Optical densities (OD)
were recorded at 450 nm using a microplate reader (model 680;
Bio-Rad Laboratories, Inc.), according to manufacturer's protocol.
Cell proliferation was plotted as OD450 compared to untreated
control cells.
Flow cytometric analysis
For cell cycle analysis, the cells were fixed with
70% ethanol overnight, treated with RNaseA (50 µg/ml;
Sigma-Aldrich) in PBS for 20 min and incubated with propidium
iodide (PI, 50 µg/ml; Sigma-Aldrich) for 30 min in the dark.
Finally, the stained cells were analyzed by fluorescence-activated
cell sorting using a flow cytometer (BD FACSCanto™ II; BD
Biosciences, Franklin Lakes, NJ, USA), and the percentage of cells
at G0/G1, S and G2/M phases were
quantified using ModFit software (ModFit LT version 3.2; BD
Biosciences). For apoptosis analysis, the cells were stained with
Annexin V and propidium iodide, using the Annexin V Apoptosis
Detection kit (BestBio), according to the manufacturer's protocol.
The rate of apoptosis was analyzed using a flow cytometer.
Statistical analysis
All the experiments were performed at least three
times using independent cell cultures of samples from a different
individual. Statistical analyses were performed using SPSS 19.0
software (SPSS IBM, Armonk, NY, USA). Data are presented as the
mean ± standard error of the mean. One-way analysis of variance was
used to determine the relationship between different groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Morphological observation and
identification of ESCs
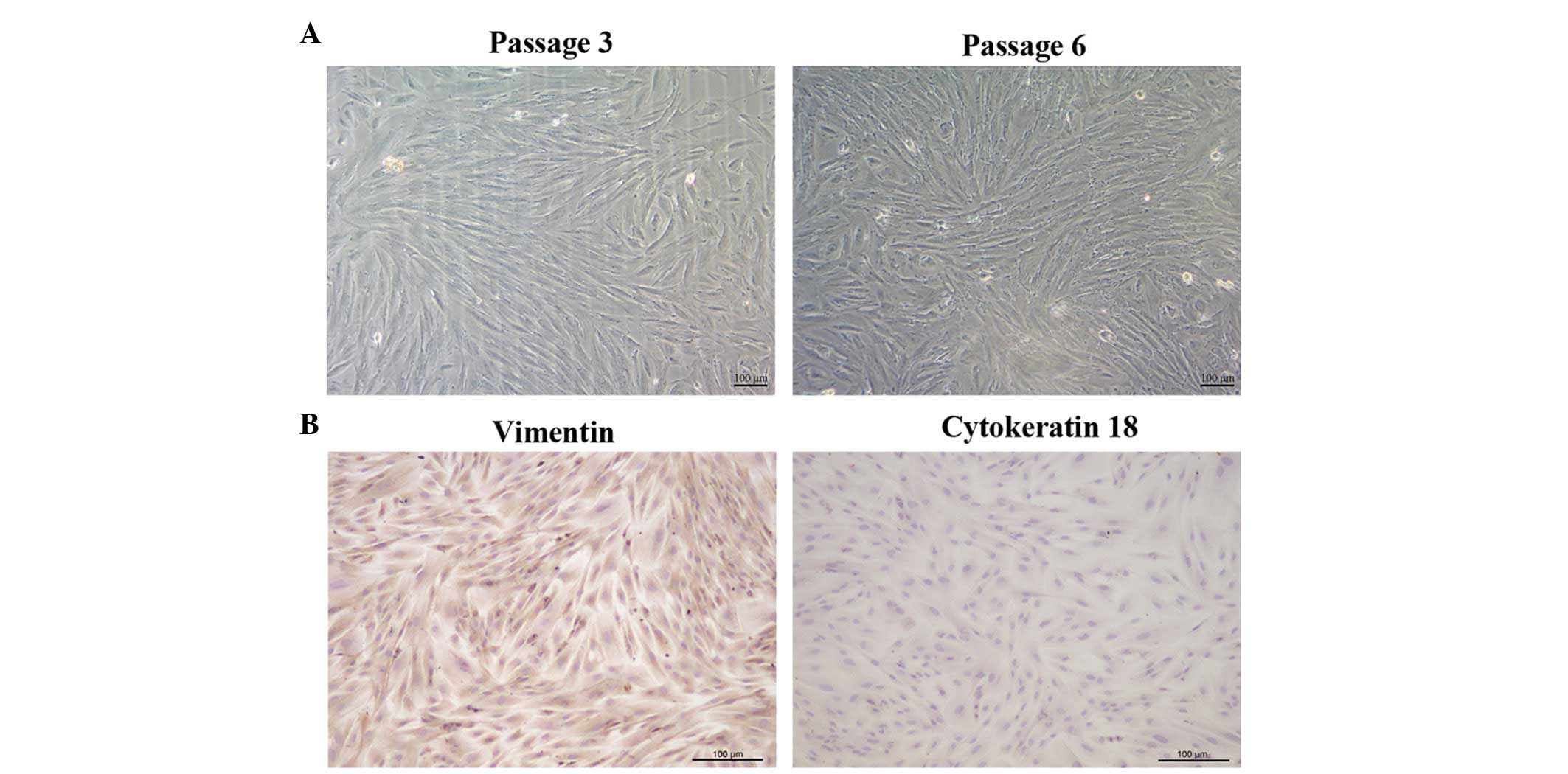
To identify and characterize fiber-like morphology
of the ESCs, immunocytochemical and microscopic observations were
performed. The ESCs were able to attach and grow 24 h after
seeding. Subsequently, ESC cultures of the 3rd and
6th generation were observed under a microscope. As
shown in Fig. 1A, the cells
exhibited a fiber-like morphology, with the majority of the ESCs
forming tightly parallel arrays. In addition, the cells were able
to grow to confluence. Immunocytochemical staining demonstrated
that the ESCs were positively stained for the stromal marker
vimentin (brown), and were negative for the epithelial cell marker
cytokeratin 18 (Fig. 1B). In
addition, the purity of the stromal cells was detected to be
>98%.
TGF-β1 increases the expression levels of
COL1A1, α-SMA and p-Smad2/3 in the ESCs and promotes
transdifferentiation of the cells
The well-known powerful pro-fibrotic effects of
TGF-β1 make it a promising candidate for the induction of a cell
model of endometrial fibrosis. In order to study the function of
TGF-β1 in primary ESCs, the cells were initially incubated with
TGF-β1 (1, 5 or 10 ng/ml) for 48 h, with vehicle (10 mM citric
acid; Peprotech, Inc.) used as a negative control. In addition, the
ESCs were incubated with TGF-β1 at 10 ng/ml for 12, 24, 48 or 72 h.
As shown in Fig. 2A and B, COL1A1
and α-SMA expression were increased in response to TGF-β1, in a
time- and dose-dependent manner. Furthermore, the addition of 1, 5
and 10 ng/ml TGF-β1 significantly increased the protein expression
levels of p-Smad2/3 in ESCs after 24, 48 and 72 h of treatment.
Notably, consistent with the phenotypic changes mentioned
previously, the primary ESCs underwent morphological changes to
become spindle-shaped myofibroblast-like cells in response to
continuous TGF-β1 stimulation for 4 days, as observed under an
electronic microscope (Fig. 2C).
These results indicate that TGF-β1 may promote myofibroblast
transdifferentiation of ESCs, activate the TGF-β1/Smad signaling
pathway and increase COL1A1 synthesis.
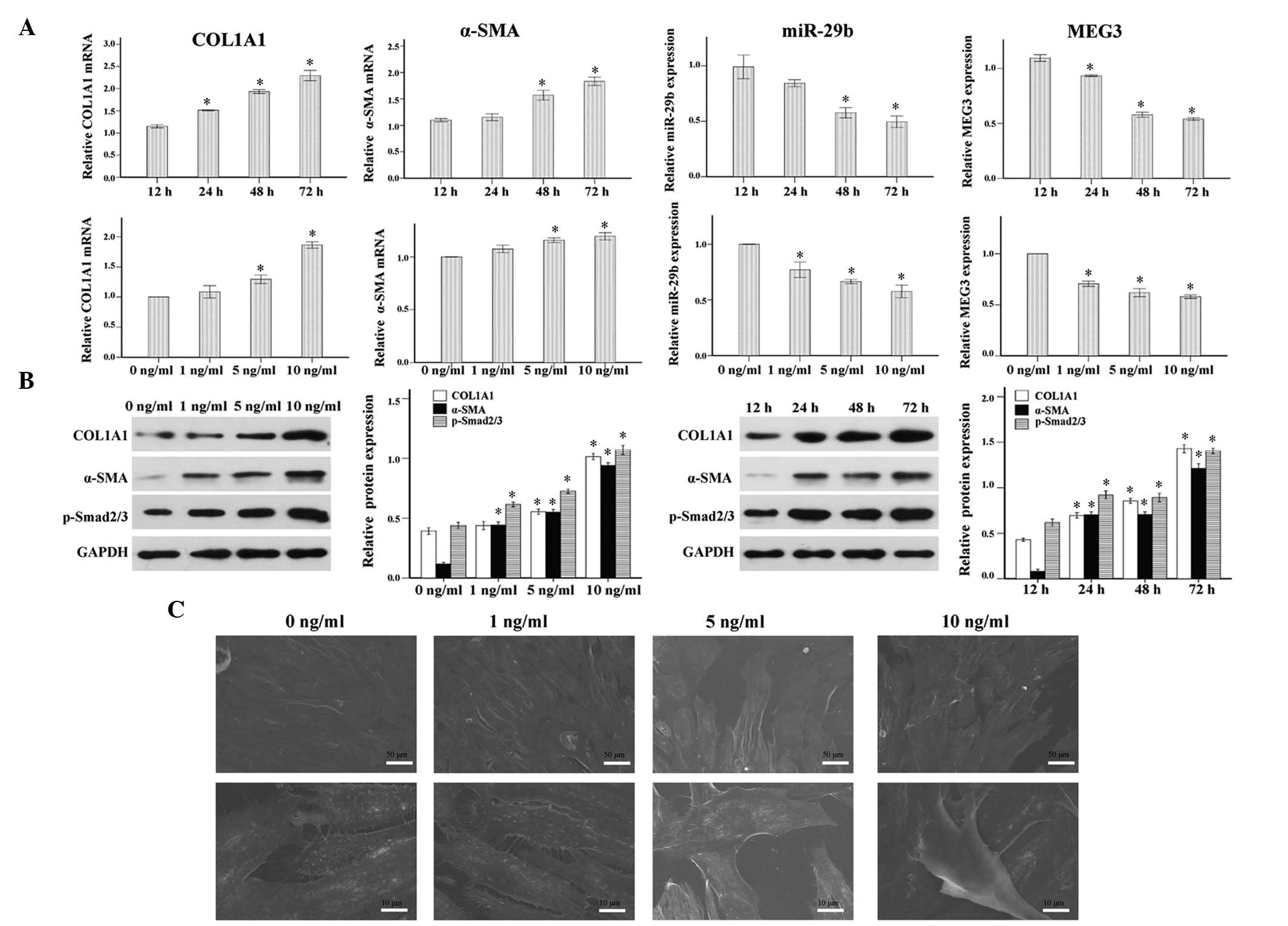 | Figure 2Pro-fibrotic effects of transforming
growth factor (TGF)-β1 in endometrial stromal cells (ESCs). Primary
ESCs were treated with TGF-β1 (0, 1, 5 or 10 ng/ml) for 48 h, or
TGF-β1 (10 ng/ml) for the indicated time (12, 24, 48 or 72 h) in
Dulbecco's modified Eagle's medium containing 1% fetal bovine
serum. (A) Time and dose-dependent effects of TGF-β1 on the mRNA
expression levels of collagen, type 1, alpha 1 (COL1A1), α-smooth
muscle actin (α-SMA), microRNA (miR)-29b and maternally expressed
gene 3 (MEG3), as analyzed by reverse transcription-quantitative
polymerase chain reaction. (B) Time and dose-dependent effects of
TGF-β1 on the protein expression levels of COL1A1, α-SMA and
p-Smad2/3 as analyzed by western blotting. (C) Scanning electron
microscopy images demonstrated the morphological changes of primary
ESCs into spindle-shaped myofibroblast-like cells in response to
continuous TGF-β1 stimulation (10 ng/ml) for 4 days. The results
are expressed as relative expression against control expression
without treatment. Data are presented as the mean ± standard error
of the mean. *P<0.05 vs. the control group (12 h or 0
ng/ml). Scale bar, 10 or 50 µm. |
miR-29b and MEG3 are downregulated in
TGF-β1-treated ESCs
To determine whether the TGF-β1/Smad pathway
interacts with miR-29b in order to mediate endometrial fibrosis,
the expression levels of miR-29b were detected following treatment
with various doses of TGF-β1 for various durations. Treatment with
TGF-β1 significantly reduced the expression levels of miR-29b in a
time- and dose-dependent manner (Fig.
2A). Notably, MEG3 was downregulated in a similar manner
following treatment of the cells with TGF-β1 (Fig. 2A).
Overexpression of miR-29b exerts
anti-fibrotic effects in ESCs
Significantly decreased miR-29b expression was
detected in TGF-β1-treated ESCs; therefore, the possible functional
role of miR-29b in ESCs activation was determined. miR-29b mimics
were transfected into ESCs before or after TGF-β1 treatment (10
ng/ml). In the ESCs that underwent preventive treatment,
overexpression of miR-29b significantly suppressed transcription
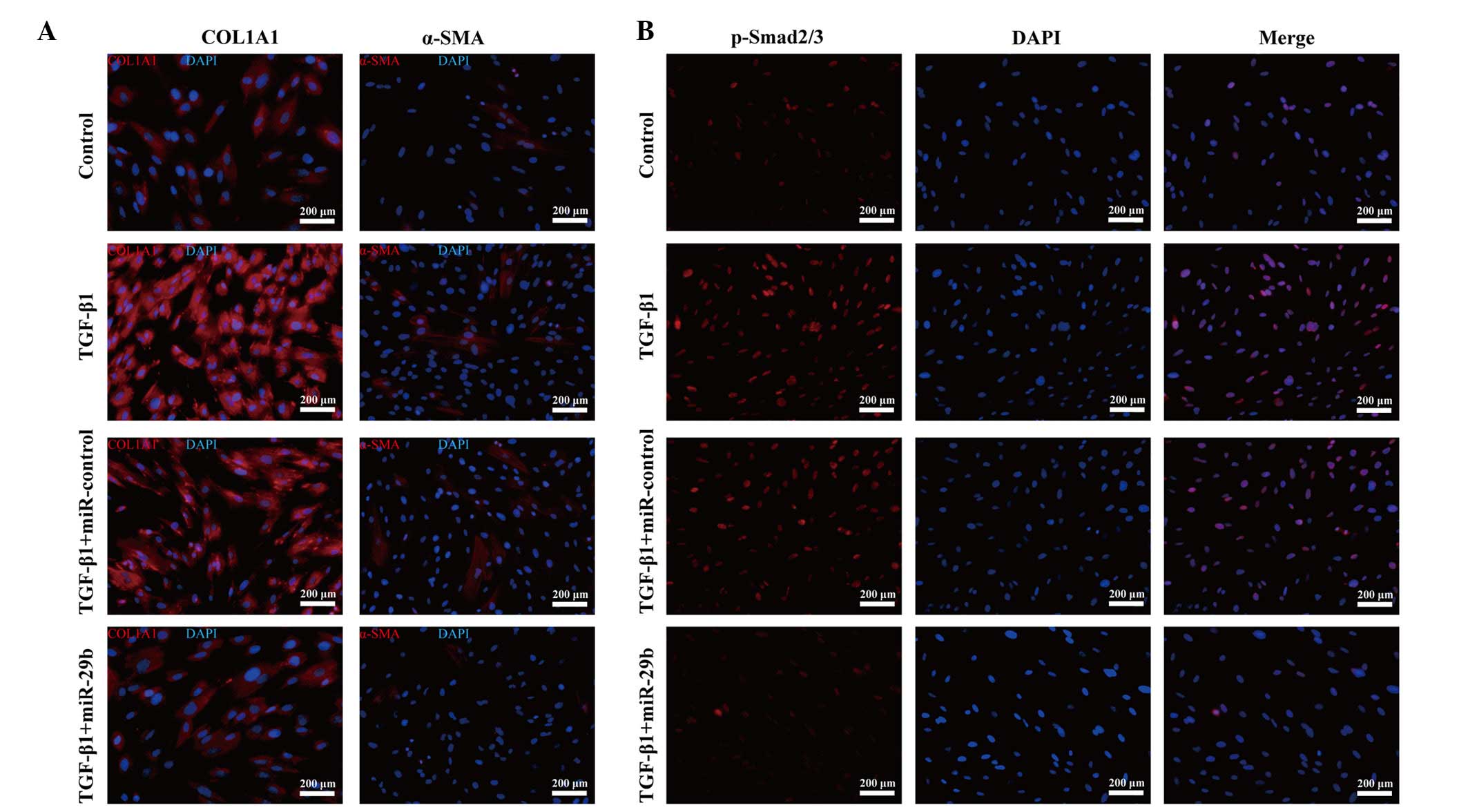
and protein synthesis of COL1A1 (Fig.
3A and B). Notably, α-SMA, an important factor of myofibroblast
transdifferentiation, was decreased post-transfection of ESCs with
miR-29b (Fig. 3A and B), although
it is not predicted to be a direct target of miR-29b by several
computational algorithms (data not shown). In the ESCs that
underwent therapeutic treatment, overexpression of miR-29b exerted
a significant anti-fibrotic effect on ESCs, via suppression of
COL1A1 expression; however, its inhibitory effects were less
marked, as compared with those induced by the preventive treatment
(Fig. 3A and C). The decreased
expression levels of COL1A1 and α-SMA were further confirmed by
immunofluorescence staining (Fig.
4A).
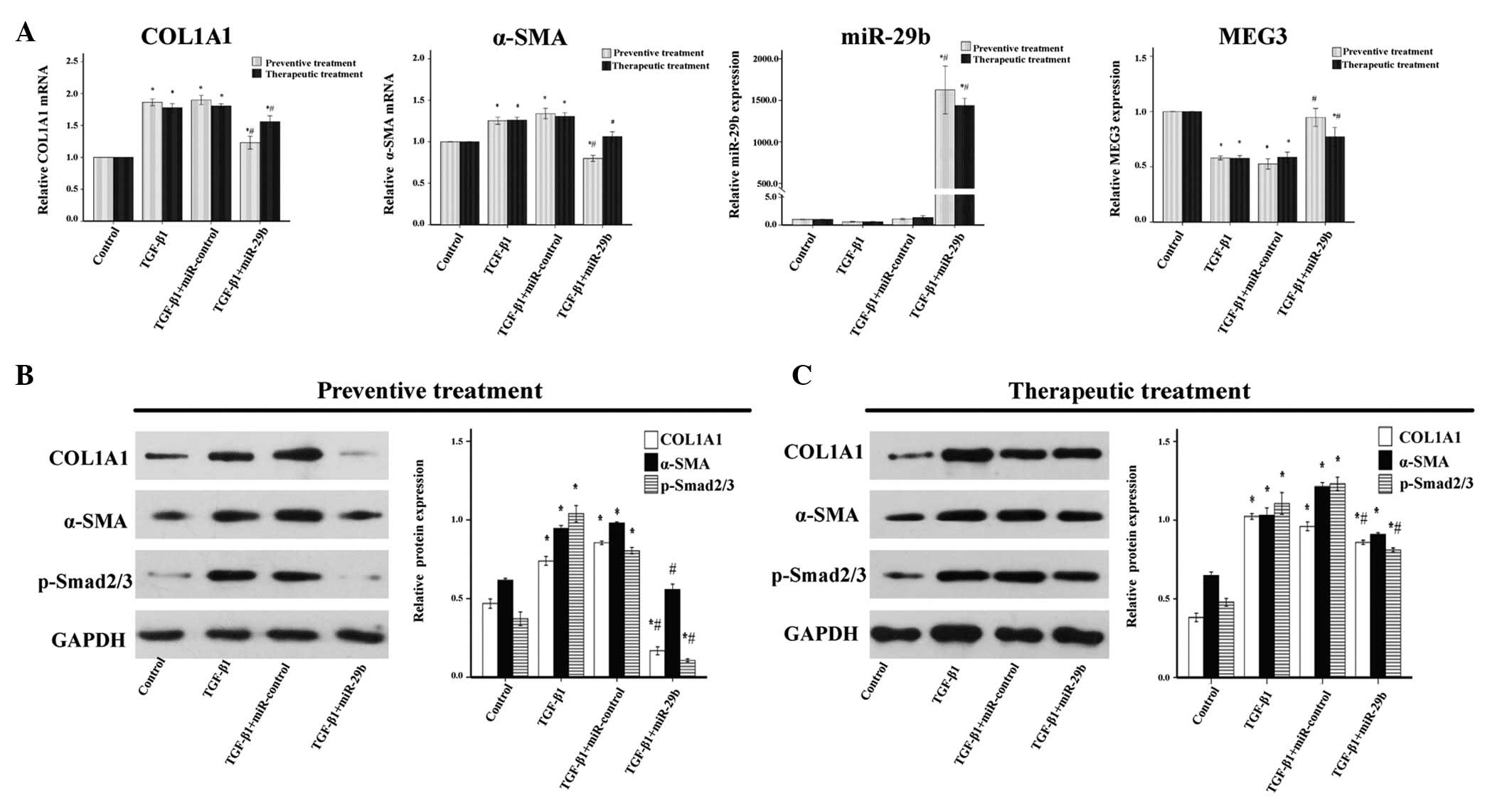 | Figure 3Anti-fibrotic effects of microRNA
(miR)-29b on transforming growth factor (TGF)-β1-treated
endometrial stromal cells (ESCs). miR-29b mimics (50 nM) were
transfected before or after TGF-β1 (10 ng/ml) treatment of ESCs.
(A) In the cells that underwent preventive treatment, exogenous
miR-29b significantly decreased the mRNA expression levels of
collagen, type 1, α 1 (COL1A1) and α-smooth muscle actin (α-SMA),
and increased the expression levels of maternally expressed 3
(MEG3), as compared with the TGF-β1-treated group, as determined by
reverse transcription-quantitative polymerase chain reaction
analysis. Similar results were observed for the therapeutic
treatment. (B) In the cells that underwent preventive treatment,
western blot analysis detected a marked downregulation of COL1A1,
α-SMA and phosphorylated (p)-Smad2/3 protein expression. (C) In the
cells that underwent therapeutic treatment, COL1A1, α-SMA and
p-Smad2/3 protein expression levels were decreased, as assessed by
western blotting in ESCs transfected with miR-29b mimics following
pretreatment with TGF-β1 for 24 h, as compared with the TGF-β1
group. Data are presented as the mean ± standard error of the mean.
*P<0.05, compared with the control group;
#P<0. 05 compared with the TGF-β1 group. GAPDH,
glyceraldehyde 3-phosphate dehydrogenase. |
miR-29b protects ESCs from fibrosis by
suppressing TGF-β1/Smad signaling
To explore the possible mechanism by which miR-29b
protects ESCs from fibrosis, apart from its direct effect on ECM
mRNAs via binding the 3′-untranslated region (UTR) (21), alterations to the TGF-β1/Smad
signaling pathway were detected post-transfection. Exogenous
overexpression of miR-29b suppressed the TGF-β1-induced
pro-fibrotic effects by inhibiting the TGF-β1/Smad signaling
pathway, as determined by a marked reduction in p-Smad2/3 protein
expression (Fig. 3B and C). In
addition, the decreased p-Smad2/3 nuclear trans-location and
accumulation detected by immunofluorescence staining supported this
theory (Fig. 4B). These results
suggest that inhibition of the TGF-β1/Smad signaling pathway may be
considered a key mechanism by which miR-29b inhibits endometrial
fibrosis in response to TGF-β1 treatment in vitro.
Overexpression of miR-29b upregulates
MEG3 in TGF-β1-stimulated ESCs and mediates suppression of cell
proliferation and promotion of cell apoptosis
To identify the alterations in MEG3 expression
post-transfection, RT-qPCR analysis was performed. Overexpression
of miR-29b increased the expression levels of MEG3 in both the
preventive and therapeutic treatment groups (Fig. 3A). Furthermore, to investigate
whether miR-29b ectopic expression had an effect on cell
proliferation and apoptosis following treatment with TGF-β1, a cell
proliferation assay, and cell cycle and apoptosis analyses were
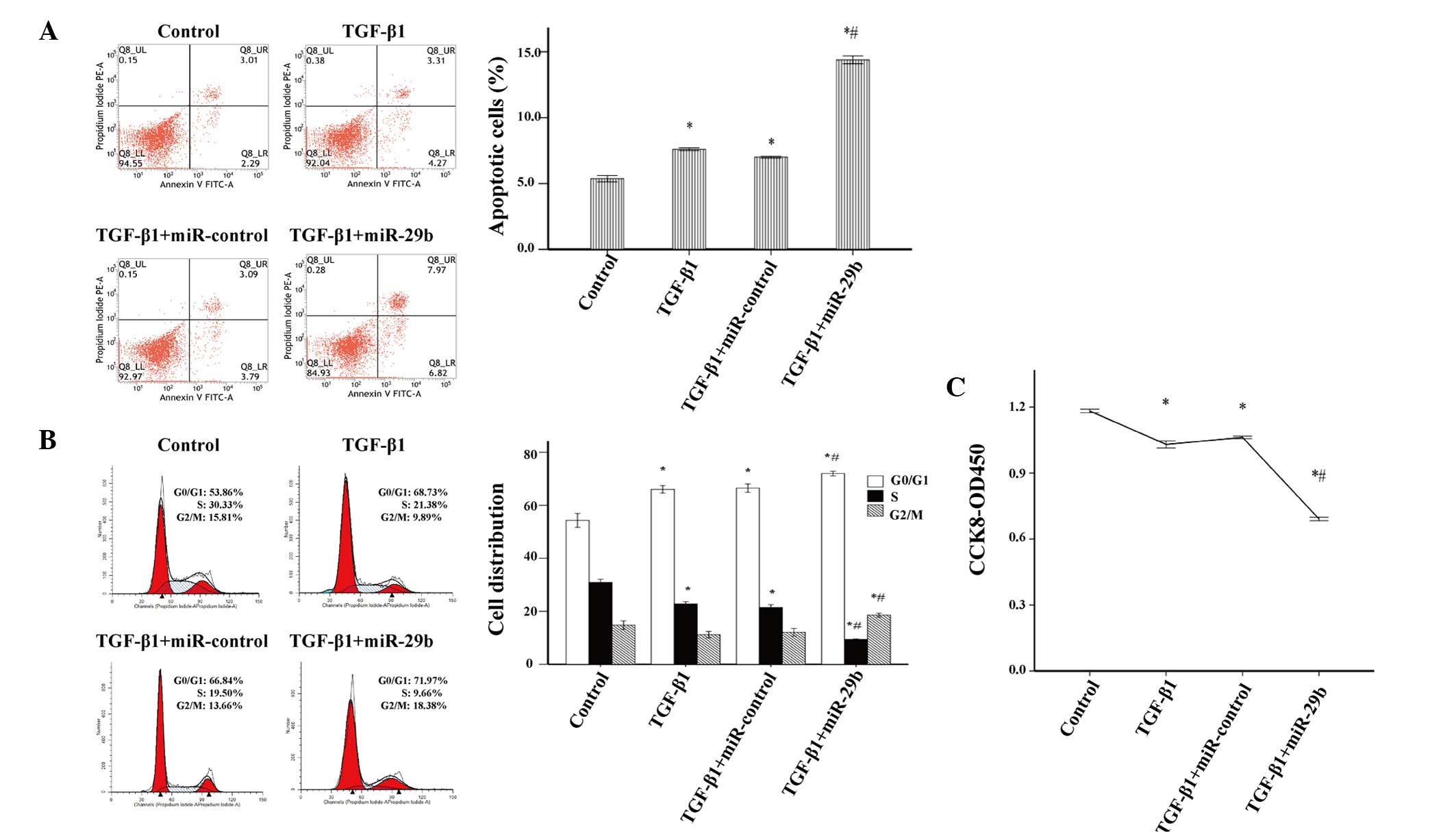
conducted. The proportion of apoptotic cells in the miR-29b
intervention group was greater than that in the miR-control group
(Fig. 5A), thus suggesting that
miR-29b may induce apoptosis in activated ESCs. In addition, the
overexpression of miR-29b significantly increased the percentage of
activated ESCs in G0/G1 and G2/M
phases, and decreased the percentage of cells in S phase (Fig. 5B). Furthermore, cell proliferation
was significantly suppressed in TGF-β1-treated ESCs transfected
with miR-29b mimics (Fig. 5C).
Discussion
TGF-β1 is recognized as an important pro-fibrogenic
mediator, which regulates ECM production and triggers myofibroblast
transition (22). The results of
the present study strongly suggested that TGF-β1 was able to exert
a significant pro-fibrotic effect on ESCs, induce myofibroblast
transdifferentiation of ESCs, activate the TGF-β1/Smad signaling
pathway and increase COL1A1 synthesis. Concordant with the findings
of previous studies, which demonstrated that TGF-β1 is a key
profibrotic factor in various cell types ranging from human hepatic
stellate cells (23) to human
conditionally immortalized podocytes (11), the results of the present study
demonstrated that TGF-β1 was able to promote fibrogenesis in ESCs.
In the present study, ESCs transdifferentiation into
myofibroblast-like cells was activated by TGF-β1, as further
verified by increased α-SMA expression, which is an important
factor contributing to the development of endometrial fibrosis.
This phenotypic transformation results in hypertrophy accompanied
by the increased secretion of ECM components and inflammatory
cytokines, which results in a vicious circle that promotes
transdifferentiation. A similar situation has been detected in the
fibrotic liver, where hepatic stellate cells constitute the main
matrix-producing cell type of the fibrotic liver following
transdifferentiation into myofibroblasts (24). In addition to the
transdifferentiation effect, TGF-β1 also induces the synthesis of
ECM components, including COL1A1 in ESCs, via the TGF-β1/Smad
signaling pathway. Similar results have been reported in various
other disease models, including post-myocardial infarction
(10), obstructive nephropathy
(25) and pulmonary fibrosis
(13). Notably, downregulation of
miR-29b and MEG3 were inversely correlated with the increased
expression levels of COL1A1 and α-SMA when the cells were treated
with TGF-β1, thus indicating the complexity of the TGF-β1-induced
fibrotic process in ESCs with the participation of mRNA, miRNA and
lncRNA.
The miR-29 family has garnered attention regarding
research into fibrotic disease in various organs, since no other
miRNA has been predicted to possess the unique characteristic of
targeting >11 of the 20 collagen genes by binding to their
3′-UTR (21). Although the
anti-fibrotic effects of miR-29 agree with the phenomenon that
TGF-β1 promotes collagen gene expression by downregulating miR-29,
it does not rule out the possibility that miR-29 modulates fibrosis
via other biological pathways. Among the miR-29 family members,
miR-29b is intriguing due to its tendency for nuclear localization
(26). Cytosolic and nuclear
miR-29b may interact with diverse mRNAs, or DNA and lncRNAs, to
elicit various biological effects. In line with previous studies
(22,24), the present study demonstrated that
loss of miR-29b in ESCs following TGF-β1 exposure led to the
increased expression of COL1A1 and α-SMA, via activation of the
TGF-β1/Smad signaling pathway. Conversely, overexpression of
miR-29b in ESCs was capable of preventing TGF-β1-induced
endometrial fibrosis in vitro via suppression of the
TGF-β1/Smad signaling pathway. In addition, the present study
detected anti-proliferative and pro-apoptotic roles for miR-29b in
activated ESCs.
In addition to the well-accepted mechanism of
miR-29b on suppression of collagen matrix expression by directly
targeting the 3′-UTR of collagen genes (10), targeting bone morphogenetic protein
1, a known activator of TGF-β1, in order to inhibit TGF-β1
transcription, may be another mechanism by which miR-29b inhibits
the pro-fibrotic effects of TGF-β1 (27). A similar recently reported
mechanism suggests that miR-29b targets the TGF-β1 coding sequence
region exon 3, thereby inhibiting the TGF-β1/Smad signaling pathway
(28). Furthermore, overexpression
of miR-29 is able to modulate DNA methyltransferase 1 and 3, and
thus increase the expression of MEG3 (15). The upregulation of MEG3 can further
induce the accumulation of p53 protein, leading to the inhibition
of cell growth (29). In the
present study, it was suggested that upregulation of MEG3 induced
by the overexpression of miR-29b may be associated with
anti-fibrotic effects in ESCs. He et al (16) reported that MEG3 inhibited cell
proliferation, increased cell apoptosis, and decreased α-SMA and
COL1A1 expression in TGF-β1-treated hepatic stellate cells. In this
respect, miR-29b may contribute to the suppression of proliferation
and promote apoptosis of activated ESCs by upregulating MEG3.
In conclusion, the present study suggested that loss
of miR-29b expression in ESCs following treatment with TGF-β1 may
lead to the transdifferentiation of ESCs into myofibroblast-like
cells, and the increased expression of COL1A1 and α-SMA, via
activation of the TGF-β1/Smad signaling pathway. Conversely,
overexpression of miR-29b efficiently overcomes the pro-fibrogenic
influence of TGF-β1 on ESCs. Overexpressing miR-29 may be
considered a promising therapeutic strategy for the treatment of
endometrial fibrosis.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China to Yuanli He (grant
no. 81270658), the Doctoral Scientific Research Foundation of
Guangzhou Medical University (grant no. 2015C27) and the Guangdong
Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene
Regulation, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen
University.
References
|
1
|
Yu D, Wong YM, Cheong Y, Xia E and Li TC:
Asherman syndrome - one century later. Fertil Steril. 89:759–779.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
March CM: Asherman's syndrome. Semin
Reprod Med. 29:83–94. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huang CC, Orvis GD, Wang Y and Behringer
RR: Stromal-to-epithelial transition during postpartum endometrial
regeneration. PLoS One. 7:e442852012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Germeyer A, Sharkey AM, Prasadajudio M,
Sherwin R, Moffett A, Bieback K, Clausmeyer S, Masters L, Popovici
RM, Hess AP, et al: Paracrine effects of uterine leucocytes on gene
expression of human uterine stromal fibroblasts. Mol Hum Reprod.
15:39–48. 2009. View Article : Google Scholar
|
|
5
|
Kamato D, Burch ML, Piva TJ, Rezaei HB,
Rostam MA, Xu S, Zheng W, Little PJ and Osman N: Transforming
growth factor-β signalling: Role and consequences of Smad linker
region phosphorylation. Cell Signal. 25:2017–2024. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Andrieux G, Fattet L, Le Borgne M, Rimokh
R and Théret N: Dynamic regulation of Tgf-B signaling by Tif1γ: A
computational approach. PLoS One. 7:e337612012. View Article : Google Scholar
|
|
7
|
Muro AF, Moretti FA, Moore BB, Yan M,
Atrasz RG, Wilke CA, Flaherty KR, Martinez FJ, Tsui JL, Sheppard D,
et al: An essential role for fibronectin extra type III domain A in
pulmonary fibrosis. Am J Respir Crit Care Med. 177:638–645. 2008.
View Article : Google Scholar
|
|
8
|
Tao Z and Duan H: Expression of
adhesion-related cytokines in the uterine fluid after transcervical
resection of adhesion. Zhonghua Fu Chan Ke Za Zhi. 47:734–737.
2012.In Chinese.
|
|
9
|
He Y, Huang C, Lin X and Li J: MicroRNA-29
family, a crucial therapeutic target for fibrosis diseases.
Biochimie. 95:1355–1359. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
van Rooij E, Sutherland LB, Thatcher JE,
DiMaio JM, Naseem RH, Marshall WS, Hill JA and Olson EN:
Dysregulation of microRNAs after myocardial infarction reveals a
role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA.
105:13027–13032. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang B, Komers R, Carew R, Winbanks CE, Xu
B, Herman-Edelstein M, Koh P, Thomas M, Jandeleit-Dahm K,
Gregorevic P, et al: Suppression of microRNA-29 expression by
TGF-β1 promotes collagen expression and renal fibrosis. J Am Soc
Nephrol. 23:252–265. 2012. View Article : Google Scholar :
|
|
12
|
Roderburg C, Urban GW, Bettermann K, Vucur
M, Zimmermann H, Schmidt S, Janssen J, Koppe C, Knolle P, Castoldi
M, et al: Micro-RNA profiling reveals a role for miR-29 in human
and murine liver fibrosis. Hepatology. 53:209–218. 2011. View Article : Google Scholar
|
|
13
|
Xiao J, Meng XM, Huang XR, Chung AC, Feng
YL, Hui DS, Yu CM, Sung JJ and Lan HY: miR-29 inhibits
bleomycin-induced pulmonary fibrosis in mice. Mol Ther.
20:1251–1260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yu JW, Duan WJ, Huang XR, Meng XM, Yu XQ
and Lan HY: MicroRNA-29b inhibits peritoneal fibrosis in a mouse
model of peritoneal dialysis. Lab Invest. 94:978–990. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Braconi C, Kogure T, Valeri N, Huang N,
Nuovo G, Costinean S, Negrini M, Miotto E, Croce CM and Patel T:
microRNA-29 can regulate expression of the long non-coding RNA gene
MEG3 in hepatocellular cancer. Oncogene. 30:4750–4756. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
He Y, Wu YT, Huang C, Meng XM, Ma TT, Wu
BM, Xu FY, Zhang L, Lv XW and Li J: Inhibitory effects of long
noncoding RNA MEG3 on hepatic stellate cells activation and liver
fibrogenesis. Biochim Biophys Acta. 1842.2204–2215. 2014.
|
|
17
|
Zhou M, He Y and Liu F: Expression and
significance of miR-29a, TGF-β1, Smad2 and Smad3 in endometrium of
patient with intrauterine adhesions. Shi Yong Yi Xue Za Zhi.
30:1231–1234. 2014.In Chinese.
|
|
18
|
Ryan IP, Schriock ED and Taylor RN:
Isolation, characterization, and comparison of human endometrial
and endometriosis cells in vitro. J Clin Endocrinol Metab.
78:642–649. 1994.PubMed/NCBI
|
|
19
|
Park DW, Choi DS, Ryu HS, Kwon HC, Joo H
and Min CK: A well-defined in vitro three-dimensional culture of
human endometrium and its applicability to endometrial cancer
invasion. Cancer Lett. 195:185–192. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Liu Y, Taylor NE, Lu L, Usa K, Cowley AW
Jr, Ferreri NR, Yeo NC and Liang M: Renal medullary microRNAs in
Dahl salt-sensitive rats: miR-29b regulates several collagens and
related genes. Hypertension. 55:974–982. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Verrecchia F and Mauviel A: Transforming
growth factor-beta and fibrosis. World J Gastroenterol.
13:3056–3062. 2007.PubMed/NCBI
|
|
23
|
Kwiecinski M, Noetel A, Elfimova N,
Trebicka J, Schievenbusch S, Strack I, Molnar L, von Brandenstein
M, Töx U, Nischt R, et al: Hepatocyte growth factor (HGF) inhibits
collagen I and IV synthesis in hepatic stellate cells by miRNA-29
induction. PLoS One. 6:e245682011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gressner AM and Weiskirchen R: Modern
pathogenetic concepts of liver fibrosis suggest stellate cells and
TGF-beta as major players and therapeutic targets. J Cell Mol Med.
10:76–99. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Qin W, Chung AC, Huang XR, Meng XM, Hui
DS, Yu CM, Sung JJ and Lan HY: TGF-β/Smad3 signaling promotes renal
fibrosis by inhibiting miR-29. J Am Soc Nephrol. 22:1462–1474.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kriegel AJ, Liu Y, Fang Y, Ding X and
Liang M: The miR-29 family: Genomics, cell biology, and relevance
to renal and cardiovascular injury. Physiol Genomics. 44:237–244.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Luna C, Li G, Qiu J, Epstein DL and
Gonzalez P: Role of miR-29b on the regulation of the extracellular
matrix in human trabecular meshwork cells under chronic oxidative
stress. Mol Vis. 15:2488–2497. 2009.PubMed/NCBI
|
|
28
|
Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM
and Lan HY: miR-29b as a therapeutic agent for angiotensin
II-induced cardiac fibrosis by targeting TGF-β/Smad3 signaling. Mol
Ther. 22:974–985. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou Y, Zhong Y, Wang Y, Zhang X, Batista
DL, Gejman R, Ansell PJ, Zhao J, Weng C and Klibanski A: Activation
of p53 by MEG3 non-coding RNA. J Biol Chem. 282:24731–24742. 2007.
View Article : Google Scholar : PubMed/NCBI
|