Introduction
Acute lymphoblastic leukemia (ALL) is a malignancy
derived from cells of the lymphoid lineage. Despite good clinical
outcomes for patients with childhood ALL, adult ALL remains
clinically challenging due to a high rate of recurrence (1). It has been previously reported that
the overall 5-year survival rate for patients with ALL recurrence
is 7% (2). Previous studies have
investigated the importance of the bone marrow microenvironment on
ALL recurrence (3).
The bone marrow microenvironment is composed of
various cell types and abundant extracellular matrix (4). Previous studies have demonstrated
that stromal cells promote the proliferation and survival of
leukemic cells by secreting soluble chemicals and adhesion
molecules, including chemokine (C-X-C motif) ligand 12, IL
(interleukin)-7 (5), vascular
endothelial growth factor A (VEGFA), platelet derived growth factor
(PDGF) (6), and via the integrin
β1/cluster of differentiation (CD)44 axis (7). Stromal cell-induced proliferation may
be associated with the development of minimal residual disease,
which is considered to be an independent prognostic factor in ALL
(8). Thus, it is necessary to
investigate the mechanisms of bone marrow microenvironment-induced
proliferation in leukemic cells. Mesenchymal stem cells (MSCs) are
important components of the solid and hematologic tumor
microenvironment (9). The present
study investigated the bone marrow microenvironment-mediated
proliferation of leukemic cells using bone marrow-derived
(BM)-MSCs.
Nuclear receptor subfamily 2 group F member 2
(NR2F2) is a member of the nuclear receptor superfamily widely
expressed in the mesenchymal compartment of developing organs
(10). NR2F2 expression was
previously reported to be abundant in MSCs and involved in the
modulation of cell differentiation (11). NR2F2 has been predominantly
investigated in solid malignancies, including ovarian and prostate
cancer (12). Whether NR2F2 is
important in the leukemic microenvironment remains to be
investigated. Notably, the present study demonstrated that NR2F2
was upregulated in BM-MSCs following co-culture with Reh cells,
which is a representative ALL cell line. The current study aimed to
investigate the importance of NR2F2 in BM-MSC-induced proliferation
of leukemic cells.
Materials and methods
Generation of BM-MSCs
Human bone marrow samples were collected from normal
donors by bone marrow aspiration at The First Affiliated Hospital,
College of Medicine, Zhejiang University (Hangzhou, China) and
mononuclear cells were isolated by density gradient centrifugation
at 400 × g at 20°C for 25 min. The current study was approved by
the First Affiliated Hospital, College of Medicine, Zhejiang
University. The cells were cultured in low-glucose Dulbecco's
modified Eagle's medium (10-014-CVR; Corning Incorporated, Corning,
NY, USA) supplemented with 10% fetal bovine serum (FBS; 10099-141;
Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) at 37°C
and 5% CO2 in a humidified incubator. The medium was
replaced after the initial 48 h. Subsequently the medium was
changed every 3 days. The adherent cells were passaged when 90%
confluence was reached. BM-MSCs at passage 2–6 were used in the
subsequent assays.
Characteristics of BM-MSCs
BM-MSCs of passages 3–5 were harvested using 0.25%
trypsin-ethylenediaminetetraacetic acid solution (EDTA; 25200-056;
Gibco; Thermo Fisher Scientific, Inc.). Cells were then incubated
with the following anti-human monoclonal antibodies:
CD90-fluorescein isothiocyanate (FITC; 1:20; 11-0909),
CD105-phycoerythrin (PE; 1:20; 12-1057), CD73-APC (allophycocyanin;
1:20; 17-0739), CD45-FITC (1:20; 11-9459), CD34-PE (1:20; 12-0349),
CD19-APC (1:20; 17-0199) and CD11b-PE (1:20; 12-0113) antibodies
(eBioscience, Inc., San Diego, CA, USA) at 4°C for 30 min.
Following fixing with 75% ethanol (Sinopharm Chemical Reagent Co.,
Ltd.), permeabilisation with permeabilisation solution [CCS012A;
Multi Sciences (Lianke) Biotech Co., Ltd., Hangzhou, China] and
blocking with Human BD Fc Block (564219; BD Pharmingen, San Diego,
CA, USA), cells were detected using an FC 500 MCL flow cytometer
(Beckman Coulter, Inc., Brea, CA, USA) with mouse IgG1 K isotype
control FITC (11-4714), mouse IgG1 K isotype control PE (12-4714),
mouse IgG1 K isotype control APC (17-4714) isotype-matched
antibodies (eBioscience, Inc.) applied as controls. BM-MSCs at
passages 3–5 were cultured in osteogenic or adipogenic
differentiation medium (HUXMA-90021 and -90031, respectively;
Cyagen Biosciences, Inc., Santa Clara, CA, USA) for the
differentiation assays. Cells were stained with Alizarin Red or Oil
red O solution (HUXMA-90031; Cyagen Biosciences, Inc.) to evaluate
osteogenic or adipogenic differentiation, respectively. The cells
were analyzed using CXP Analysis Software 5 (Beckman Coulter,
Inc.).
Cell culture and co-culture
The human ALL Reh cells were obtained from the Cell
Bank of Shanghai Institute of Biochemistry and Cell Biology,
Chinese Academy of Sciences (Shanghai, China), and were divided
into cells cultured either alone or co-cultured with with BM-MSCs.
Reh cells were cultured in Iscove's modified Dulbecco's medium
(SH30228; Hyclone; GE Healthcare Life Sciences, Logan, UT, USA)
supplemented with 10% FBS at 37°C and 5% CO2 in a
humidified incubator. For co-culture experiments, BM-MSCs were
initially seeded on 6- or 12-well plates at 5×104
cells/ml density. After 4–6 h of culture, Reh cells were added to
the adherent MSCs layer at 5×105 cells/ml density. After
48 h of co-culture, Reh cells were collected by directly separating
suspending cells as described by previous studies (13,14).
Ki-67 detection by flow cytometry
Forkhead Box P3/Transcription Factor Staining Buffer
Set (eBioscience, Inc.) was used for intracellular staining with
the anti-human monoclonal Ki-67-PE antibody (12-5699; 1:20;
eBioscience, Inc.). Reh cells were collected and washed with
phosphate-buffered saline (PBS) 2 times. Following incubation with
fixation/permeabilization solution at 4°C for 30 min, Reh cells
were washed and incubated with 5 µl Ki-67-PE antibody at 4°C
for 30 min. Cells were resuspended in 500 µl PBS for flow
cytometric detection using an FC 500 MCL flow cytometer. Ki-67 was
detectable during the active phases of cell cycle (G1,
S, G2 and M) and not during the resting phase
(G0), therefore, the positive cells were considered to
be proliferating cells (15).
Apoptosis assay
Reh cells were harvested and washed twice with cold
PBS. Cells were resuspended in 100 µl 1X binding buffer
provided in the Apoptosis Detection kit (559763; BD Pharmingen, San
Diego, CA, USA). Following incubation with the Annexin V-PE
antibody and 7-aminoactinomycin D from the Apoptosis Detection kit
(559763; BD Pharmingen) at room temperature for 15 min, Reh cells
were analyzed using the FC 500 MCL flow cytometer. Cells that were
Annexin V-positive were considered to be apoptotic.
Cell cycle assay
Reh cells were fixed with chilled 75% ethanol for 2
h at −20°C. Following rehydration in PBS for 15 min, cells were
washed twice and resuspended in 1 ml propidium iodide (CCS012A;
Multi Sciences Biotech Co., Ltd., Hangzhou, China), then incubated
at room temperature for 30 min. Samples were analyzed using the FC
500 MCL flow cytometer. Data were analyzed using MultiCycle
software, version 3.2 (Beckman Coulter, Inc.).
Knock-down of NR2F2
A previously reported (16), NR2F2-targeting shRNA
(5′-AGGTAACGTGATTGATTCAGTATCTTA-3′) was cloned into the pGLV3
plasmid (Genepharm, Inc., Sunnyvale, CA, USA). A scrambled sequence
(5′-TTCTCCGAACGTGTCACGT-3′) was also cloned into the pGLV3 as a
negative control. shRNAs were obtained from Shanghai GenePharma
Co., Ltd. (Shanghai, China). Highly concentrated lentivirus was
produced by co-transfecting 293T cells (Cell Bank of Shanghai
Institute of Biochemistry and Cell Biology, Chinese Academy of
Sciences) with pMD2.G (Thermo Fisher Scientific, Inc.), psPAX2
(Thermo Fisher Scientific, Inc.) and shRNA plasmids with Lipofectin
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. The viral suspension was collected, filtered and used
to transfect BM-MSCs directly following removal of medium. After 10
h, virus-containing medium was replaced with fresh medium. The
positive expression of green fluorescent protein was assessed by
fluorescent microscopy (Eclipse-Ti K11301; Nikon Corporation,
Tokyo, Japan) and used to evaluate transfection efficiency.
Cell viability assay
Transfected BM-MSCs were seeded onto a 96-well plate
at a concentration of 5×103 cells/well. Five parallel
wells of cells were seeded for each group (knockdown BM-MSCs group
and negative control BM-MSCs group). Following incubation for 48 h,
10 µl 3-(4,5-dimethyl-2-thiazolyl) 2,5-diphe-nyltetrazolium
bromide (MTT; LK-MTT001; Hangzhou Lianke Biology Technology Ltd.,
Co., Hangzhou, China) was added to all wells. Medium was discarded
after 4 h. The optical density (OD) values were evaluated by the
SpectraMax M5 microplate reader (Molecular Devices, LLC, Sunnyvale,
CA, USA) at 570 nm following addition of 100 µl dimethyl
sulfoxide (Sinopharm Chemical Reagent Co., Ltd., Shanghai,
China).
Enzyme-linked immunosorbent assay
(ELISA)
The concentration of VEGFA in the co-culture medium
was assessed using an ELISA kit (BMS277/2; eBioscience, Inc.).
Briefly, co-culture medium supernatant was collected at 24 or 48 h
following separation of Reh cells by concentration using
centrifugation at 300 × g at 20°C for 10 min. An adequate volume of
sample was incubated with pre-coated plates according to the
manufacturer's protocol. The OD value was detected at 450 nm using
the SpectraMax M5 microplate reader. Standard curves were produced
using the standards of known concentration supplied in the kit and
used to calculate protein concentrations.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Co-cultured BM-MSCs were isolated by thoroughly
washing with PBS until all Reh cells were removed. Total cellular
RNA was extracted from co-cultured BM-MSCs using TRIzol reagent
(15596-026; Thermo Fisher Scientific, Inc.) PrimeScript RT reagent
kit with gDNA Eraser (RR047A; Takara Biotechnology Co., Ltd.,
Dalian, China) was used for RT. The RT protocol was as follows:
37°C for 15 min; 85°C for 5 sec; stored at 4°C. qPCR was performed
using SYBR Premix Ex Taq II (RR420A; Takara Biotechnology Co.,
Ltd.) according to manufacturer's instructions in a LightCycler
system (Roche Diagnostics, Basel, Switzerland). Each reaction was
performed in triplicate. The cycling conditions were as follows:
Initial denaturation at 95°C for 5 min, 45 cycles of 95°C for 5 sec
and 60°C for 20 sec, melting curve analysis at 95°C for 15 sec,
65°C for 15 sec and then maintained at 95°C. The expression of
glyceraldehyde 3-phophate dehydrogenase (GAPDH) was used as a
reference gene. The primers used were as follows: NR2F2, forward
5′-CAACCAGCCGACGAGATT-3′ and reverse
5′-ATTGCTCTATGACTGAGGAGGAGAC-3′; VEGFA, forward
5′-ACAGGTACAGGGATGAGGACAC-3′ and reverse
5′-AAGCAGGTGAGAGTAAGCGAAG-3′; GAPDH, forward
5′-AGAAGGCTGGGGCTCATTTG-3′ and reverse
5′-AGGGGCCATCCACAGTCTTC-3′.
Western blot analysis
The transfected BM-MSCs were harvested by digesting
using 0.25% trypsin-EDTA solution. Cells were lysed on ice for 1 h
in radioimmunoprecipitation assay lysis buffer (AR0105; Wuhan
Boster Biological Technology, Ltd., Wuhan, China) with
phenylmethylsulfonyl fluoride solution (AR1179; Wuhan Boster
Biological Technology, Ltd.). Cell lysates were centrifuged at
12,000 × g for 5 min at 4°C. The supernatant was incubated at 100°C
for 10 min following addition of an appropriate volume of loading
buffer (5-fold dilution; Hangzhou Fu De Biological Technology, Co.,
Ltd., Hangzhou, China). Samples were loaded onto a 10%
polyacrylamide gels containing sodium dodecyl sulfate (Hangzhou Fu
De Biological Technology, Co., Ltd.), electrophoretically separated
(110 V for 100 min) and transferred onto a nitrocellulose membrane.
Membranes were blocked at 20°C for 2 h with 5% non-fat milk then
incubated with the primary antibodies at 4°C overnight, and then
incubated with fluorescent IgG IRDye® 650 goat
anti-mouse (1:10,000; 926-65010) and goat anti-rabbit (1:10,000;
926-65020) secondary antibodies (LI-COR, Inc., Lincoln, NE, USA) at
room temperature for 1 h. Rabbit monoclonal NR2F2 (1:1,000; #6434)
and mouse monoclonal GAPDH (1:1,000; #5079) primary antibodies were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA)
and Multi Sciences (Lianke) Biotech Co., Ltd., respectively.
Immunoreactive bands were visualized using an Odyssey Infrared
Imaging System (LI-COR, Inc.). GAPDH was used as a loading
control.
Statistical analysis
All independent experiments were repeated ≥3 times.
Statistical analysis was performed using SPSS software, version
19.0 (IBM SPSS, Armonk, NY, USA). Differences between mean values
were evaluated using Student's two-tailed t-test. Values are
presented as the mean ± standard deviation and P<0.05 was
considered to indicate a statistically significant difference.
Results
Characteristics of BM-MSCs
BM-MSCs exhibited a spindle-like shape and
fibroblast-like morphology. BM-MSCs were CD90- and CD105-positive,
and predominantly CD73-positive. However, the cells were negative
for CD19 and CD11b, and hematopoietic markers, including CD34 and
CD45. The characteristics of BM-MSCs have been described in a
previous report (17).
BM-MSCs promote the proliferation of Reh
cells
In order to assess whether BM-MSCs promote Reh cell
proliferation, Reh cells were isolated and the expression of the
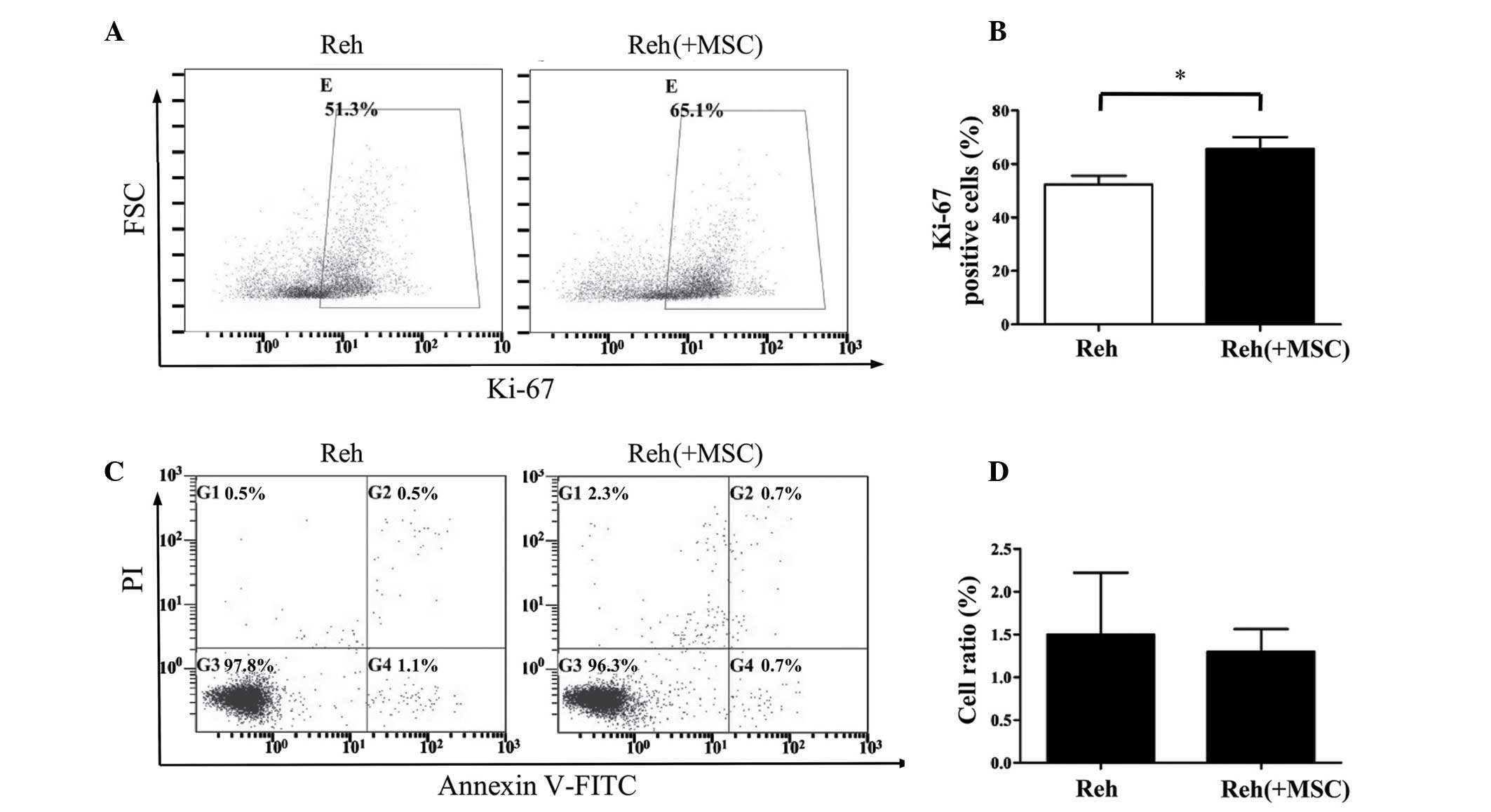
Ki-67 antigen was analyzed. As demonstrated in Fig. 1A and B, Reh cells that were
co-cultured with BM-MSCs for 48 h exhibited a significantly
increased percentage of Ki-67-positive cells compared with Reh
cells cultured alone (P<0.05). The present study also assessed
whether BM-MSCs affected the apoptosis of Reh cells. Co-culture
with BM-MSCs for 48 h had no observable effect on the apoptosis of
Reh cells (Fig. 1C and D).
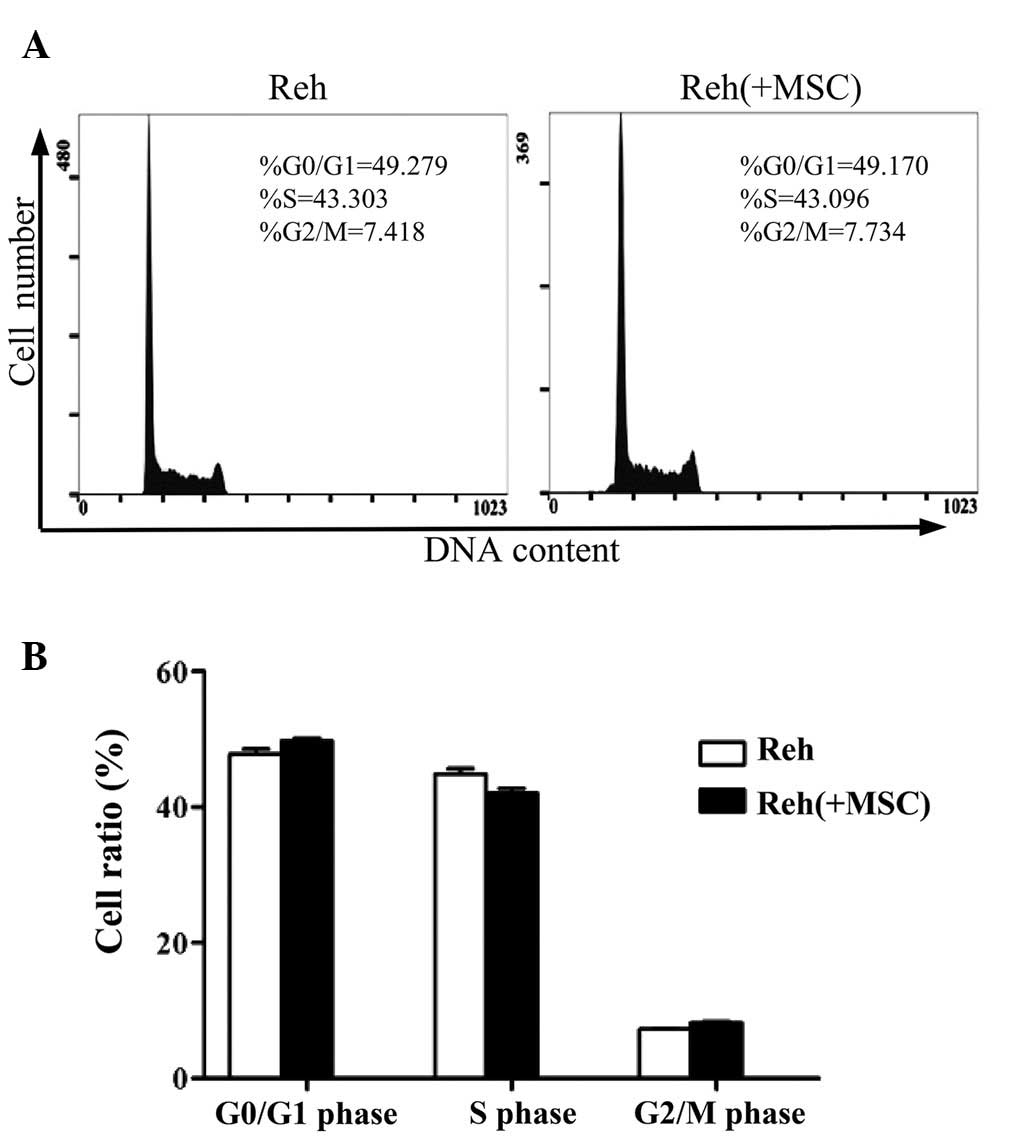
Additionally, the cell cycle pattern of Reh cells was analyzed in
the co-culture system. When co-cultured with BM-MSCs, the
percentage of Reh cells in the G2/M phase was markedly
increased compared with Reh cells cultured alone. Whereas the
percentages of cells in G0/G1 or S phases
were not significantly different (Fig.
2).
NR2F2 regulates the BM-MSCs-promoted
proliferation of Reh cells
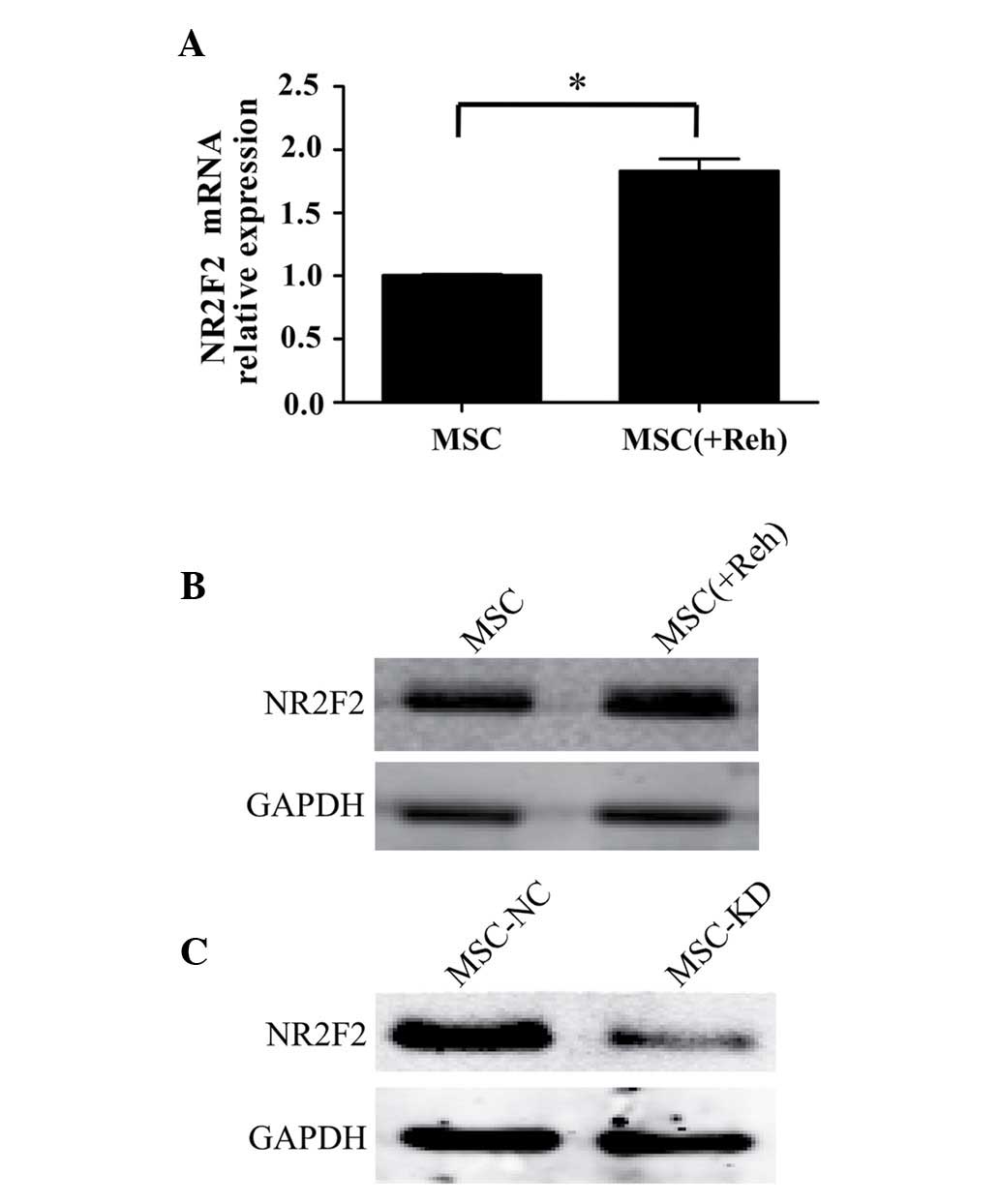
To analyze the expression levels of NR2F2 in
BM-MSCs, BM-MSCs were separated from Reh cells following co-culture
for 48 h. The mRNA and protein levels of NR2F2 in BM-MSCs were
observed to be upregulated when co-cultured with Reh cells compared
with culture alone (Fig. 3A and
B). In order to further elucidate whether NR2F2 is involved in
mediating the BM-MSC-induced proliferation of Reh cells, NR2F2
expression was measured in BM-MSCs, and was identified to be
downregulated by transfection of an NR2F2 antisense shRNA. Western
blot analysis demonstrated that the expression of NR2F2 was
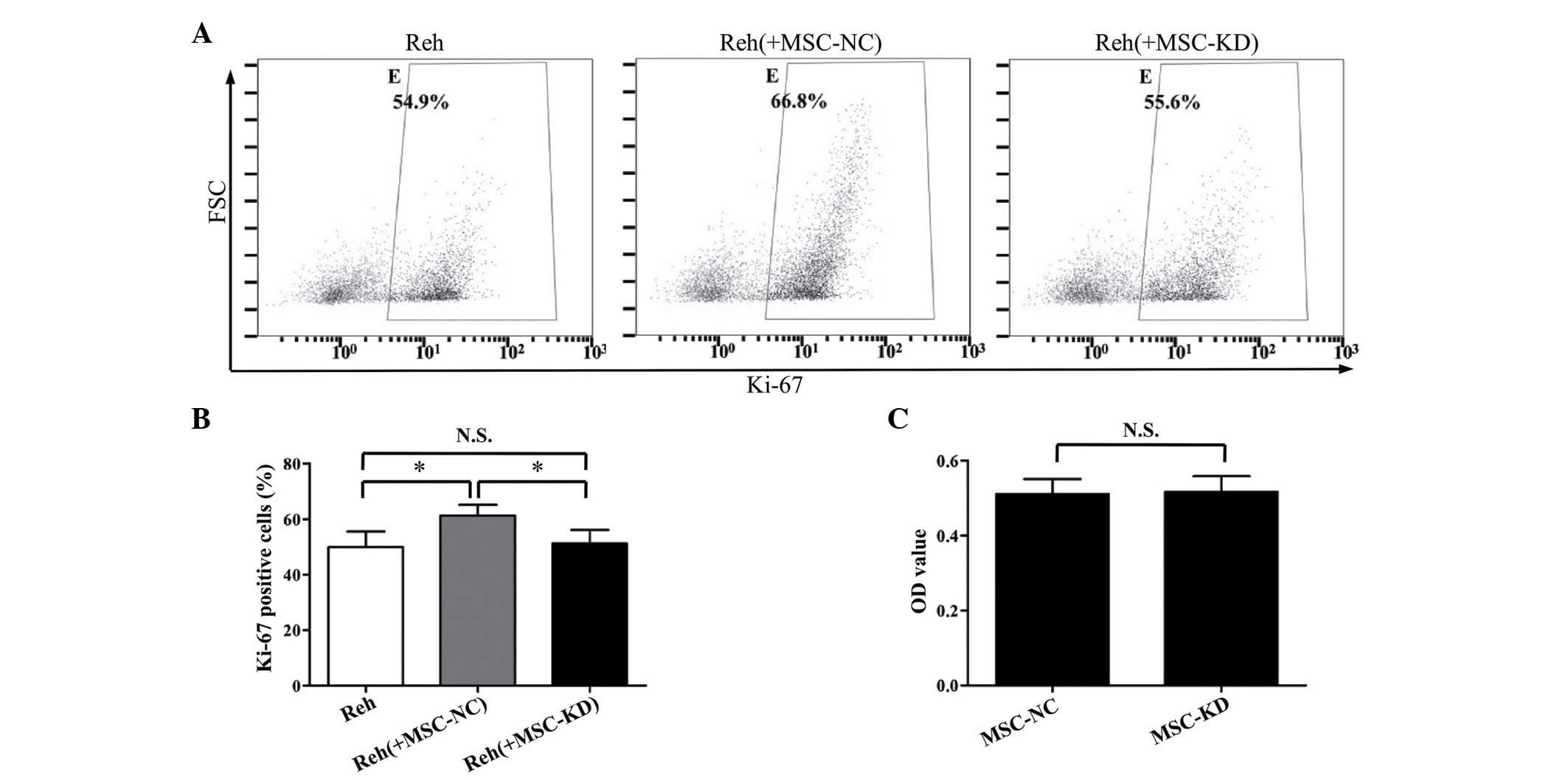
significantly knocked down by the shRNA (Fig. 3C). As demonstrated in Fig. 4A and B, the percentage of
Ki-67-positive Reh cells in the NR2F2 knockdown BM-MSC co-culture
group was significantly reduced compared with the negative control
group (P<0.05). Furthermore, the percentage of Ki-67-positive
Reh cells in the NR2F2 knockdown BM-MSC co-culture group was
similar to the percentage observed when Reh cells were cultured
alone. Additionally, the cell viabilities of BM-MSCs in the
knockdown and control groups were compared to exclude the
possibility that silencing NR2F2 affected the cell viability. As
demonstrated by the MTT assay, no significant difference in cell
viability was observed between the two groups (Fig. 4C). These data suggested that NR2F2
is involved in the regulation of BM-MSC-induced proliferation of
Reh cells.
Downregulation of NR2F2 suppresses the
expression of VEGFA in BM-MSCs
As it has been previously reported that VEGFA
provides important proliferative cues for ALL cells in a
BM-MSC-conditioned system (6), the
present study attempted to evaluate whether the effects of NR2F2 on
the co-culture system were mediated by VEGFA. BM-MSCs were isolated
from the co-culture after 48 h and the mRNA expression levels of
VEGFA were measured in NR2F2 knockdown and negative control cells.
Following NR2F2 knockdown, VEGFA was significantly downregulated
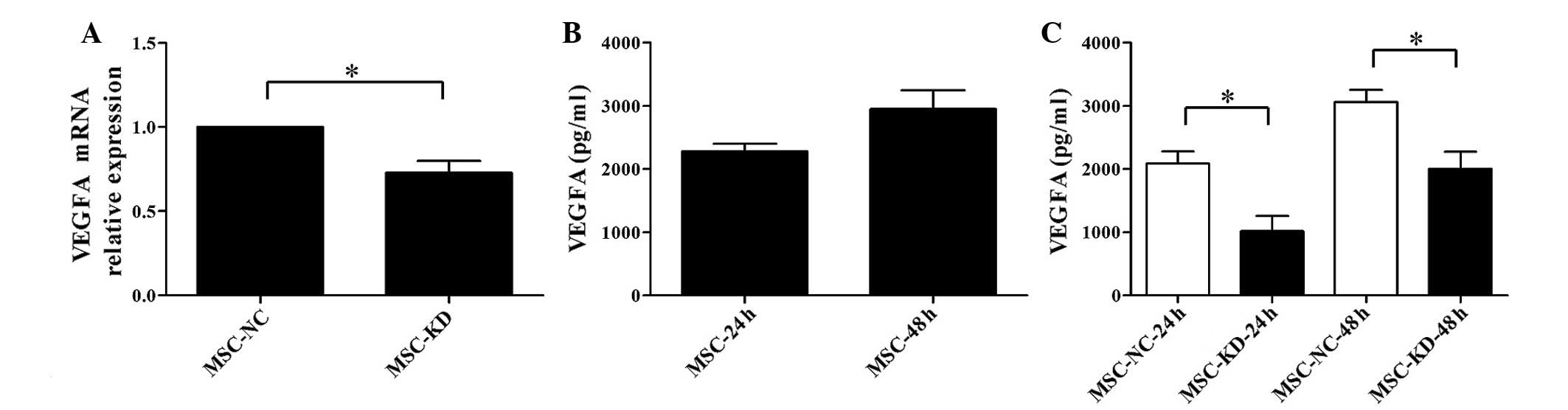
(P<0.05; Fig. 5A). To further
investigate the association between NR2F2 and VEGFA, the
concentration of VEGFA in the co-culture supernatant was measured
by ELISA at 24 and 48 h. A relatively high concentration of VEGFA
was detected in the supernatant of BM-MSCs (Fig. 5B). However, the concentration of
VEGFA was barely detectable in the supernatant of Reh cells
cultured alone (data not shown). The concentration of VEGFA was
significantly reduced in the NR2F2-knockdown group compared with
the negative control at 24 and 48 h (P<0.05; Fig. 5C). Thus, these results demonstrated
that VEGFA is involved in the mediation of the effects of NR2F2 on
BM-MSC-induced proliferation of Reh cells.
Discussion
The proliferation and survival of ALL cells has been
previously reported to be enhanced by stromal cells (6). The present study demonstrated that
following co-culture with BM-MSCs, the percentage of Ki-67
antigen-positive Reh cells was increased, demonstrating that
BM-MSCs induced Reh cell proliferation. Additionally, in accordance
with a previous study (18),
apoptosis was measured and no significant differences were observed
between Reh cells cultured alone or those co-cultured with BM-MSCs.
Yang et al (13) reported
that MSCs induce the proliferation of Reh cells and demonstrated
that the percentage of Reh cells in the S and G2/M
phases was increased in co-culture conditions. The present study
only observed an increased percentage of Reh cells in
G2/M phase, no observable changes in the percentage of
cells in S phase were detected. Although the data of the present
study is different to that of a previous study (13), this may be explained by the fact
that the cell cycle assay used in the present study did not
distinguish between the G0 and G1 phases.
Previous studies have demonstrated that stromal
cell-mediated increases in proliferation are primarily caused by
cytokines produced by stromal cells, including IL-6, VEGFA and PDGF
(6). However, the mechanisms
involved in producing these cytokines remain unclear. The data of
the present study indicated that NR2F2 levels are upregulated in
BM-MSCs co-cultured with Reh cells. Furthermore, downregulation of
NR2F2 with shRNA attenuated BM-MSC-induced proliferation of Reh
cells, which demonstrates that NR2F2 is involved in this process.
The present study also demonstrated that knockdown of NR2F2 did not
affect the viability of BM-MSCs after 48 h culture. Therefore, the
mechanism of NR2F2-regulated BM-MSC-induced proliferation is not
associated with effects on cell viability.
NR2F2 has been previously reported to regulate
VEGF/VEGF receptor 2 signaling in endothelial cells (19), thus, the present study hypothesized
that NR2F2 participates in the regulation of VEGFA in the
co-culture system. The results of RT-qPCR and ELISA assays
demonstrated that downregulation of NR2F2 in BM-MSCs inhibited the
production of VEGFA. Further research is required to explore the
specific mechanisms that mediate the effect of NR2F2 on the
production of VEGFA in BM-MSCs. The present study additionally
demonstrated that VEGFA is secreted by BM-MSCs and detected in the
culture supernatant. However, VEGFA was not detected in the
supernatant of Reh cells cultured alone. These data suggest that
BM-MSC-induced proliferation of Reh cells is partially mediated by
the secretion of VEGFA.
In conclusion, the present study demonstrated that
BM-MSCs promote the proliferation of Reh cells and that this
process was partially mediated by NR2F2 regulation of VEGFA
expression. Targeting NR2F2 may be a potential therapeutic strategy
to inhibit the proliferation of ALL cells by disrupting the
microenvironmental support.
Acknowledgments
The current study was supported by the National
Natural Science Foundation of China (grant nos. 81230014, 81100364
and 81370647) and the Natural Science Foundation of Zhejiang
Province (grant no. Y13H080008).
References
|
1
|
Faderl S, O'Brien S, Pui CH, Stock W,
Wetzler M, Hoelzer D and Kantarjian HM: Adult acute lymphoblastic
leukemia: Concepts and strategies. Cancer. 116:1165–1176. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fielding AK, Richards SM, Chopra R,
Lazarus HM, Litzow MR, Buck G, Durrant IJ, Luger SM, Marks DI,
Franklin IM, et al Medical Research Council: of the United Kingdom
Adult ALL Working Party; Eastern Cooperative Oncology Group:
Outcome of 609 adults after relapse of acute lymphoblastic leukemia
(ALL); an MRC UKALL12/ECOG 2993 study. Blood. 109:944–950. 2007.
View Article : Google Scholar
|
|
3
|
Shishido S, Bönig H and Kim YM: Role of
integrin alpha4 in drug resistance of leukemia. Front Oncol.
4:992014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Clark BR and Keating A: Biology of bone
marrow stroma. Ann N Y Acad Sci. 770:70–78. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Juarez J, Baraz R, Gaundar S, Bradstock K
and Bendall L: Interaction of interleukin-7 and interleukin-3 with
the CXCL12-induced proliferation of B-cell progenitor acute
lymphoblastic leukemia. Haematologica. 92:450–459. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gaundar SS, Bradstock KF and Bendall LJ:
p38MAPK inhibitors attenuate cytokine production by bone marrow
stromal cells and reduce stroma-mediated proliferation of acute
lymphoblastic leukemia cells. Cell Cycle. 8:2975–2983. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Malfuson JV, Boutin L, Clay D, Thépenier
C, Desterke C, Torossian F, Guerton B, Anginot A, de Revel T,
Lataillade JJ and Le Bousse-Kerdilès MC: SP/drug efflux
functionality of hematopoietic progenitors is controlled by
mesenchymal niche through VLA-4/CD44 axis. Leukemia. 28:853–864.
2014. View Article : Google Scholar
|
|
8
|
van Dongen JJ, van der Velden VH,
Brüggemann M and Orfao A: Minimal residual disease diagnostics in
acute lymphoblastic leukemia: Need for sensitive, fast, and
standardized technologies. Blood. 125:3996–4009. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Casey SC, Vaccari M, Al-Mulla F,
Al-Temaimi R, Amedei A, Barcellos-Hoff MH, Brown DG, Chapellier M,
Christopher J, Curran CS, et al: The effect of environmental
chemicals on the tumor microenvironment. Carcinogenesis. 36(Suppl
1): S160–S183. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pereira FA, Qiu Y, Zhou G, Tsai MJ and
Tsai SY: The orphan nuclear receptor COUP-TFII is required for
angiogenesis and heart development. Genes Dev. 13:1037–1049. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xie X, Qin J, Lin SH, Tsai SY and Tsai MJ:
Nuclear receptor chicken ovalbumin upstream promoter-transcription
factor II (COUP-TFII) modulates mesenchymal cell commitment and
differentiation. Proc Natl Acad Sci USA. 108:14843–14848. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Safe S, Jin UH, Hedrick E, Reeder A and
Lee SO: Minireview: Role of orphan nuclear receptors in cancer and
potential as drug targets. Mol Endocrinol. 28:157–172. 2014.
View Article : Google Scholar :
|
|
13
|
Yang Y, Mallampati S, Sun B, Zhang J, Kim
SB, Lee JS, Gong Y, Cai Z and Sun X: Wnt pathway contributes to the
protection by bone marrow stromal cells of acute lymphoblastic
leukemia cells and is a potential therapeutic target. Cancer Lett.
333:9–17. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hu K, Gu Y, Lou L, Liu L, Hu Y, Wang B,
Luo Y, Shi J, Yu X and Huang H: Galectin-3 mediates bone marrow
microenvironment-induced drug resistance in acute leukemia cells
via Wnt/β-catenin signaling pathway. J Hematol Oncol. 8:12015.
View Article : Google Scholar
|
|
15
|
Beresford MJ, Wilson GD and Makris A:
Measuring proliferation in breast cancer: Practicalities and
applications. Breast Cancer Res. 8:2162006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li L, Xie X, Qin J, Jeha GS, Saha PK, Yan
J, Haueter CM, Chan L, Tsai SY and Tsai MJ: The nuclear orphan
receptor COUP-TFII plays an essential role in adipogenesis, glucose
homeostasis, and energy metabolism. Cell Metab. 9:77–87. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang B, Hu Y, Liu L, Hu K, Tie R, He Y, Fu
S, Zhu N, Luo Y, Yu X and Huang H: Phenotypical and functional
characterization of bone marrow mesenchymal stem cells in patients
with chronic graft-versus-host disease. Biol Blood Marrow
Transplant. 21:1020–1028. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang Y, Hu K, Hu Y, Liu L, Wang B and
Huang H: Bone marrow mesenchymal stromal cells affect the cell
cycle arrest effect of genotoxic agents on acute lymphocytic
leukemia cells via p21 down-regulation. Ann Hematol. 93:1499–1508.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen X, Qin J, Cheng CM, Tsai MJ and Tsai
SY: COUP-TFII is a major regulator of cell cycle and Notch
signaling pathways. Mol Endocrinol. 26:1268–1277. 2012. View Article : Google Scholar : PubMed/NCBI
|