Introduction
Nemaline myopathy (NM) is a genetically and
clinically heterogeneous form of myopathy, which has an incidence
rate of ~1/50,000 live births worldwide (1). In China, <50 cases, as determined
by clinical analyses and pathohistological examinations, have been
reported in the literature since the first case in 1990 (2–6). In
a previous study, in 4,127 patients with suspected myopathy who
received muscle biopsies, the occurrence of NM was only ~0.29% (28
cases) (2), thus suggesting that
NM is a rare disorder in Chinese populations. To date, at least
eight genes, including nebulin (NEB), skeletal muscle
α-actin 1 (ACTA1), cofilin-2, troponin T1, β-tropomyosin,
slow-muscle α-tropomyosin, kelch repeat and BTB domain-containing
13 and kelch-like family member 40, have been reported to cause NM
(7). The majority of these genes
encode sarcomeric thin filament proteins. Among these causal genes,
~50% of the autosomal recessive mutations associated with NM were
attributed to the NEB gene, whereas 20% of the associated
autosomal dominant or autosomal recessive mutations were attributed
to the ACTA1 gene (7). To
gain insight into the pathogenesis of NM, several animal models,
including mouse (8) and zebrafish
(9), have been established, and
various therapeutic approaches are under investigation (10).
In addition to clinical studies regarding NM,
mechanistic studies regarding molecular and genetic pathogenesis
have been conducted (11).
However, only two compound heterozygous variants (c.9052 G>A,
c.24579G>A) in the NEB gene have been identified to date
in two patients with NM in a Chinese family (6). No mutations in the other
aforementioned genes, to the best of our knowledge, have been
detected in patients with NM in China. Although genetic testing is
recommended in patients with congenital myopathy, its clinical
application has been limited by the cost and time-consuming nature
of conventional sequencing. The present study adapted the targeted
exome capture technique for rapid sequencing of potential mutations
in a patient with congenital myopathy. The present study identified
a de novo missense mutation in ACTA1 in a Chinese Han
patient with a milder NM phenotype, and also compared the clinical
manifestations in previously reported cases with the same
mutation.
Materials and methods
Patient information
The patient was an 11-year-old girl presenting with
muscle weakness, she was the second child born to a healthy
nonconsanguineous 24-year-old mother and 27-year-old father. There
was no family history of neuromuscular or other inherited diseases.
Creatine kinase measurements and electromyography assessment were
performed routinely in the hospital. Written informed consent was
obtained from the patient's parents. The present study was approved
by the ethics committee of Wenzhou Medical University (Wenzhou,
China).
Targeted gene exome sequencing
Genomic DNA was isolated from the peripheral blood
leukocytes of the patient and her parents using Wizard®
Genomic DNA Purification kit (Promega Corporation, Madison, WI,
USA). The BestSeq target-capture sequencing panel, which contains
125 known causal genes for inherited muscle diseases, was used to
analyze the DNA of the patient and her parents (conducted by
Huakang Gene Institute, Beijing, China). Polymerase chain reaction
and Sanger sequencing using primers for the ACAT1 N117S mutation
(forward, CGCGTAGCCCTCATAAATGG; reverse, CGACGAGGCTCAGAGCAAGA) were
also performed by Huakang Gene Institute to confirm the
mutation.
Results
The patient was found to have delayed gross motor
skills. She could walk independently at two years old and continued
to exhibit progressive improvement in motor development during
childhood. Her fine motor activities were adequate; however, her
knee jerk reaction and ankle reflex could not be obtained. In
addition, her biceps and triceps reflexes could not be obtained. No
positive pyramidal sign was detected. The patient had difficulty
walking upstairs, and could not jump. No sensory disturbance was
identified. Her intellectual capacity was equivalent to that of an
11-year-old child. Her height and body weight were in the normal
range. Furthermore, an electromyogram indicated possible myogenic
damage. Her serum creatine kinase level was 25 U/l, which is
slightly lower than the normal reference range (26-140 U/l).
The clinical manifestations of this patient
suggested an undiagnosed myopathy. To identify potential
mutation(s), either a de novo mutation or a recessive
mutation associated with muscular diseases, BestSeq target-capture
sequencing was conducted using DNA samples from the patient and her
parents. In the panel of 125 known causal genes for inherited
muscle diseases, a missense mutation in exon 3 of the ACTA1
gene at chromosome 1q42.13 (NM_001100.3) was identified. In
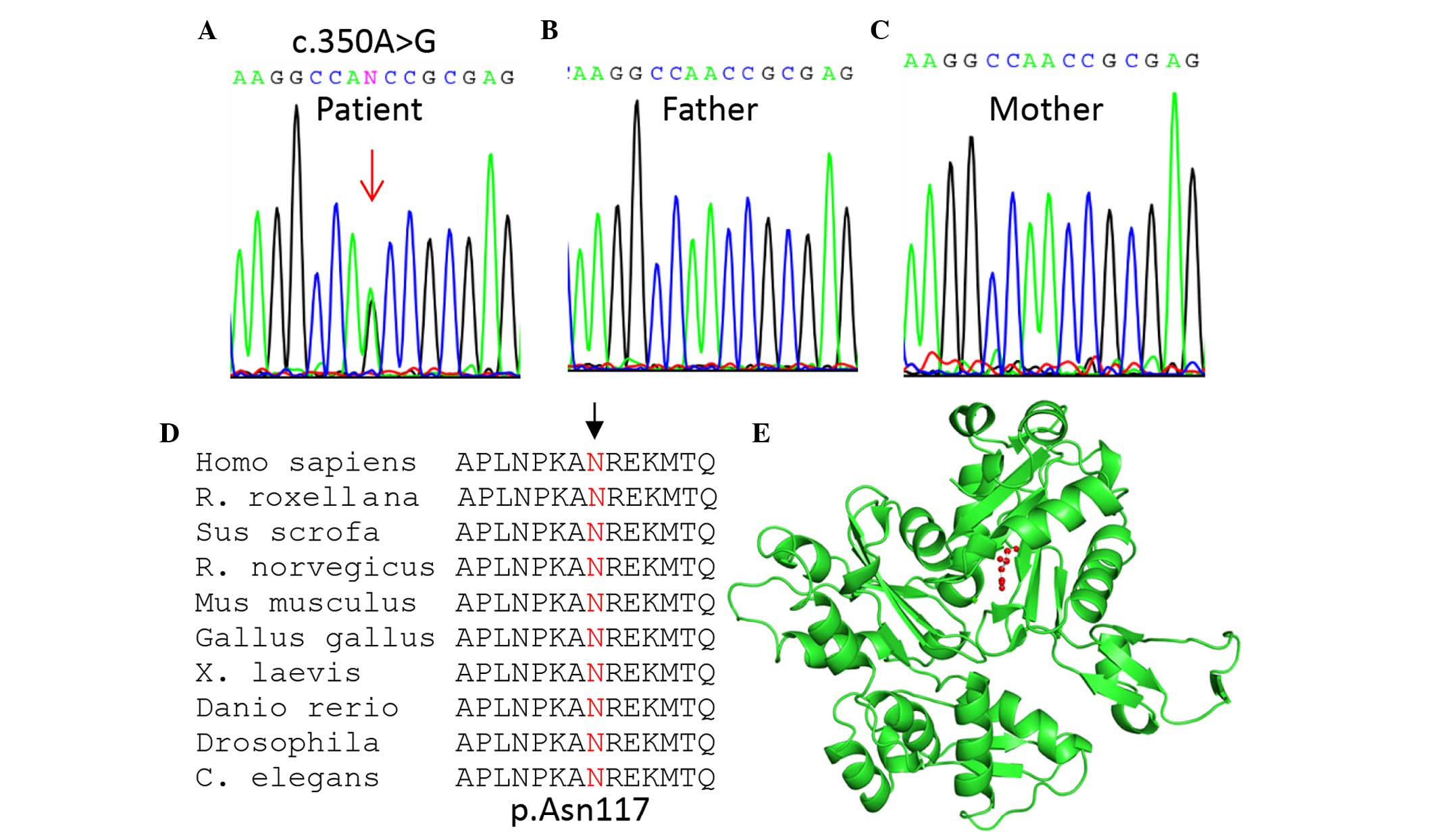
addition, the mutation (c.350A>G; p.Asn117Ser) was confirmed to
be a de novo mutation since it was only detected in the
patient (Fig. 1A), and not her
parents (Fig. 1B and C).
The identified mutation was predicted to be damaging
or disease-causing using three commonly used tools: SIFT
(http://sift.jcvi.org/; affecting protein function
with a score of 0.00), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/; possibly
damaging, with a score of 0.921), and MutationTaster (http://www.mutationtaster.org/; disease-causing
with a probability score of 0.99999). The N117 residue (as
indicated by the arrow; Fig. 1D)
is located in the nucleotide-binding domain of the sugar
kinase/heat shock protein 70/actin superfamily, which is
evolutionarily conserved from human to Caenorhabditis
elegans as shown by multiple sequence alignment (Fig. 1D). Based upon three-dimensional
structural analysis using PyMOL Molecular Graphics system, version
1.3 (www.pymol.org), N117 (designated as N115 since
the first two amino acids were removed in the crystal structure)
was located near the nucleotide-binding cleft and was thus
predicted to affect nucleotide binding (Fig. 1E).
Notably, the same mutation (p.N117S) in the
ACTA1 gene has previously been identified as a pathogenic
locus (rs121909520) in Australian patients with an autosomal
dominant inheritance pattern of NM (12–14),
and was associated with a typical or severe phenotype (Table I). The three affected individuals
succumbed at 36, 18, and 3 years of age, respectively. Conversely,
the patient presented in the current study exhibited a much milder
phenotype (Table I), thus
suggesting a genetic heterogeneity or the existence of genetic
modifiers.
 | Table IPhenotypic comparison of patients with
the N117S skeletal muscle α-actin 1 mutation. |
Table I
Phenotypic comparison of patients with
the N117S skeletal muscle α-actin 1 mutation.
| Author (year) | Patient | Age | Type of mutation | Phenotype | Reference |
|---|
| Nowak et al
(1999) | #7 (mother), family
6 | 33 (deceased) | Autosomal
dominant | Typical NM | (12) |
| #8 (child), family
6 | 18 (deceased) | Autosomal
dominant | Typical NM | (12) |
| #9 (child), family
6 | 3 (deceased) | Autosomal
dominant | Severe NM | (12) |
| Yang et al
(2016) | Proband (sporadic
case) | 11 (alive) | De novo
dominant | Mild NM could walk
freely, could not jump | Present study |
Discussion
NM (OMIM, #161800) is a novel form of congenital
myopathy that was described in 1963 (15), and is characterized by abnormal
thread- or rod-like structures in muscle fibers upon histological
examination. In the Human Gene Mutation Database professional
database (http://www.hgmd.cf.ac.uk/ac/index.php), a total of 196
mutations in the ACTA1 gene, including 181 missense
mutations, 10 indels and five splicing events, have been curated.
ACTA1 mutations (OMIM, *102610) have been reported to induce
several subtypes of myopathy, including nemaline myopathy 3 and
congenital myopathy with excess of thin myofilaments or cores
(OMIM, #161800), and congenital myopathy with fiber-type
disproportion 1 (OMIM, #255310).
In general, two clinical groups of NM can be readily
distinguished: i) Typical NM is the most common form (46% of all NM
cases), presenting as infantile hypotonia and muscle weakness; and
ii) severe NM, which is observed in 16% of NM cases, and is
characterized by an absence of spontaneous movement or respiration
at birth, arthrogryposis, and mortality in the first months of life
(16). Mutations in the
ACTA1 gene account for ~20% of typical NM cases and ~50% of
severe NM cases (16). In the
present study, NM was derived from a de novo mutation, and
the patient presented with non-progressive muscle weakness.
Compared with previously reported patients with the same mutation,
the phenotype of the patient described in the present study appears
much milder.
The N117S mutation is mostly associated with milder,
typical NM since it is located near the nucleotide-binding cleft,
and is predicted to affect nucleotide binding, a defect that may
result in a decreased polymerization propensity (14). It has also been observed that the
N117S mutant was not readily integrated into stress fibers, since
the mutant produced a diffuse cytoplasmic staining in the majority
of undifferentiated myoblasts (17). In addition, compared with the
wild-type gene, the N117S mutant has been reported to have reduced
copolymerization capacity with wild-type actin (18).
In conclusion, to the best of our knowledge, the
present study is the first to identify a pathogenic mutation in
ACTA1 in a Chinese patient with suspected myopathy. The
present study benefited from exome-target capture and
next-generation sequencing technology, as recently applied in NM
and other inherited diseases (19–22).
Further application of this approach will be helpful for the
identification of several undiagnosed patients with this type of
highly heterogeneous disorder.
Acknowledgments
The present study was supported by research funding
from the Chinese Ministry of Health Project (grant no. 201302002 to
X.C) and Wenzhou Medical University Research Funding (to P.Y.).
References
|
1
|
Wallgren-Pettersson C, Kääriäinen H,
Rapola J, Salmi T, Jääskeläinen J and Donner M: Genetics of
congenital nemaline myopathy: A study of 10 families. J Med Genet.
27:480–487. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yin X, Pu CQ, Wang Q, Liu JX and Mao YL:
Clinical and pathological features of patients with nemaline
myopathy. Mol Med Rep. 10:175–182. 2014.PubMed/NCBI
|
|
3
|
Han Y, Zheng H and Ding S: Delayed onset
of nemaline myopathy: A case report. Chin Med J (Engl).
116:798–800. 2003.
|
|
4
|
Jiang H, Xiao B, Jia DD, Zhang N, Xu XP
and Tang BS: Clinical and pathologic analysis of an autosomal
recessive kindred with nemaline myopathy. Zhonghua Yi Xue Za Zhi.
89:3316–3319. 2009.In Chinese.
|
|
5
|
Jiang C, Wang J and Lu H: Clinical and
pathological features of childhood-onset nemaline myopathy: A
report of four cases. Case Rep Med. 2012:2036022012.PubMed/NCBI
|
|
6
|
Chen Z, Wang JL, Tang BS, Sun ZF, Shi YT,
Shen L, Lei LF, Wei XM, Xiao JJ, Hu ZM, et al: Using
next-generation sequencing as a genetic diagnostic tool in rare
autosomal recessive neurologic Mendelian disorders. Neurobiol
Aging. 34:2442.e11–e17. 2013. View Article : Google Scholar
|
|
7
|
Romero NB, Sandaradura SA and Clarke NF:
Recent advances in nemaline myopathy. Curr Opin Neurol. 26:519–526.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Garg A, O'Rourke J, Long C, Doering J,
Ravenscroft G, Bezprozvannaya S, Nelson BR, Beetz N, Li L, Chen S,
et al: KLHL40 deficiency destabilizes thin filament proteins and
promotes nemaline myopathy. J Clin Invest. 124:3529–3539. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sztal TE, Zhao M, Williams C, Oorschot V,
Parslow AC, Giousoh A, Yuen M, Hall TE, Costin A, Ramm G, et al:
Zebrafish models for nemaline myopathy reveal a spectrum of
nemaline bodies contributing to reduced muscle function. Acta
Neuropathol. 130:389–406. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wallgren-Pettersson C, Sewry CA, Nowak KJ
and Laing NG: Nemaline myopathies. Semin Pediatr Neurol.
18:230–238. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tian L, Ding S, You Y, Li TR, Liu Y, Wu X,
Sun L and Xu T: Leiomodin-3-deficient mice display nemaline
myopathy with fast-myofiber atrophy. Dis Model Mech. 8:635–641.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nowak KJ, Wattanasirichaigoon D, Goebel
HH, Wilce M, Pelin K, Donner K, Jacob RL, Hübner C, Oexle K,
Anderson JR, et al: Mutations in the skeletal muscle alpha-actin
gene in patients with actin myopathy and nemaline myopathy. Nat
Genet. 23:208–212. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ilkovski B, Cooper ST, Nowak K, Ryan MM,
Yang N, Schnell C, Durling HJ, Roddick LG, Wilkinson I, Kornberg
AJ, et al: Nemaline myopathy caused by mutations in the muscle
alpha-skeletal-actin gene. Am J Hum Genet. 68:1333–1343. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sparrow JC, Nowak KJ, Durling HJ, Beggs
AH, Wallgren-Pettersson C, Romero N, Nonaka I and Laing NG: Muscle
disease caused by mutations in the skeletal muscle alpha-actin gene
(ACTA1). Neuromuscul Disord. 13:519–531. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shy GM, Engel WK, Somers JE and Wanko T:
Nemaline Myopathy. A new congenital myopathy. Brain. 86:793–810.
1963. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
North KN and Ryan MM: Nemaline myopathy.
GeneReviews® [Internet]. Pagon RA, Adam MP, Ardinger HH,
Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC,
Smith RJH and Stephens K: University of Washington; Seattle, WA:
1993–2016
|
|
17
|
Bathe FS, Rommelaere H and Machesky LM:
Phenotypes of myopathy-related actin mutants in differentiated
C2C12 myotubes. BMC Cell Biol. 8:22007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Costa CF, Rommelaere H, Waterschoot D,
Sethi KK, Nowak KJ, Laing NG, Ampe C and Machesky LM: Myopathy
mutations in alpha-skeletal-muscle actin cause a range of molecular
defects. J Cell Sci. 117:3367–3377. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sambuughin N, Zvaritch E, Kraeva N, Sizova
O, Sivak E, Dickson K, Weglinski M, Capacchione J, Muldoon S, Riazi
S, et al: Exome analysis identifies Brody myopathy in a family
diagnosed with malignant hyperthermia susceptibility. Mol Genet
Genomic Med. 2:472–483. 2014. View
Article : Google Scholar
|
|
20
|
Langouet M, Siquier-Pernet K, Sanquer S,
Bole-Feysot C, Nitschke P, Boddaert N, Munnich A, Mancini GM,
Barouki R, Amiel J and Colleaux L: Contiguous mutation syndrome in
the era of high-throughput sequencing. Mol Genet Genomic Med.
3:215–220. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu P, Cui Y, Cai W, Wu H, Xiao X, Shao Q,
Ma L, Guo S, Wu N, Jin ZB, et al: Lysosomal storage disease in the
brain: Mutations of the β-mannosidase gene identified in autosomal
dominant nystagmus. Genet Med. 17:971–979. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cai T, Yang L, Cai W, Guo S, Yu P, Li J,
Hu X, Yan M, Shao Q, Jin Y, et al: Dysplastic spondylolysis is
caused by mutations in the diastrophic dysplasia sulfate
transporter gene. Proc Natl Acad Sci USA. 112:8064–8069. 2015.
View Article : Google Scholar : PubMed/NCBI
|