Introduction
Gastric cancer (GC) is the fifth most common type of
malignancy worldwide and is the third leading cause of
cancer-related mortality (1).
Worldwide, 70% of GC cases occur in developing countries and half
in Eastern Asia, predominantly in China (1). As the majority of GC cases are
clinically advanced at diagnosis, surgery is no longer possible,
thus chemotherapy is essential (2). Cisplatin is the most common
chemotherapeutic agent for GC (3).
Although the combination of cisplatin and S-1 is an established
first-line chemotherapeutic strategy for advanced GC (4,5),
intrinsic and acquired resistance to cisplatin are major obstacles
to effective treatment. Understanding the underlying mechanisms of
cisplatin resistance in GC is critical to overcoming it.
The phosphatidylinositol 3-kinase (PI3K)/AKT
signaling pathway regulates a number of aspects of cancer
progression (6,7) and is also considered to be a
multidrug resistance locus in cancer (8). In GC, AKT and phosphorylated AKT
(p-AKT) are detected in 74 and 78% of tumors, respectively
(9). Overexpression of AKT
decreases GC sensitivity to cisplatin (10,11).
AKT activation contributes to acquired cisplatin resistance in GC
(12,13). Hypoxia is another cause of GC
resistance to cisplatin therapy (13–15).
Cells in solid tumors are subjected to a range of low oxygen
conditions and varying hypoxic intensities based on local oxygen
diffusion (16). Hypoxia-inducible
factor (HIF)-1α is a fundamental driver of cellular adaptation to
hypoxia (14,17). Reportedly, the PI3K/AKT pathway
activation upregulates HIF-1α expression (13,18,19),
which contributes to cisplatin resistance in GC cells (13,14).
It was therefore suggested that AKT may be central to intrinsic and
acquired cisplatin resistance in GC.
Bufalin, a traditional Chinese medicine, has been
shown to exhibit anticancer activity (20). Our previous study demonstrated that
bufalin markedly inhibits proliferation and promotes apoptosis
through endoplasmic reticulum stress in hepatocellular carcinoma
cells (21). Experimental studies
have shown that bufalin affects cancer cells by inhibiting the AKT
pathway (22–25). The present study demonstrated that
intrinsic and acquired resistance to cisplatin can be diminished by
bufalin, which affects the AKT pathway in GC cells.
Materials and methods
Cell culture
SGC7901, MKN-45 and BGC823 human GC cells were
obtained from the Chinese Academy of Sciences Cell Bank (Shanghai,
China). The cells were routinely cultured in RPMI-1640 medium
(Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal
calf serum (Sigma-Aldrich) at 37°C in a humidified atmosphere of 5%
CO2. To induce hypoxia, cells were incubated in a
hypoxia chamber containing 1% O2, 5% CO2 and
94% N2 at 37°C.
Antibodies and reagents
The rabbit anti-human monoclonal antibodies (Abs)
against AKT (#2920; 1:2,000), p-AKT (Ser473; #4060; 1:2,000),
glycogen synthase kinase (GSK)-3β (#12456; 1:1,000), phosphorylated
GSK3β (p-GSK3β; Ser9; #5558; 1:1,000), mammalian target of
rapamycin (mTOR; #2983; 1:1,000), phosphorylated mTOR (p-mTOR;
Ser2448; #5536; 1:1,000), eukaryotic translation initiation factor
4E binding protein 1 (4EBP1; #9644; 1:1,000), phosphorylated 4EBP1
(P-4EBP1; Ser65; #9451; 1:1,000), ribosomal protein S6 kinase (S6K;
#5707; 1:1,000) and phosphorylated S6K (p-S6K; Thr389; #9206;
1:1,000) were obtained from Cell Signaling Technology, Inc.
(Shanghai, China). Anti-β-actin (mouse IgG1; sc-47778; 1:2,000) and
anti-HIF-1α (rabbit IgG; sc-10790; 1:500) were obtained from Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Cisplatin and
bufalin were purchased from Sigma-Aldrich. GDC0068 and Fumonisin B1
were from Santa Cruz Biotechnology, Inc. The reagents were
dissolved in 100% dimethyl sulfoxide (DMSO; Sigma-Aldrich) and were
diluted with Dulbecco's modified Eagle's medium (Sigma-Aldrich) to
the desired concentrations; all concentrations contained a final
DMSO concentration of less than 0.1%.
Establishment of SGC7901
cisplatin-resistant cells
SGC7901 cisplatin-resistant cells were developed by
continuous exposure to cisplatin starting at 0.5 µM and
increasing in a stepwise manner to 5 µM. Cells that were
able to survive in the medium containing 5 µM cisplatin were
considered to be cisplatin-resistant and termed SGC7901-CR
(13,26).
Cell viability and apoptosis assays
SGC7901, MKN-45, BGC823 cells were randomly divided
into different groups. Each group of cells was cultured under
normoxic or hypoxic conditions, then were treated with 0, 50, 100
or 200 nmol/l bufalin for 48 h, or were treated with 0.10 µM
cisplatin and/or 0.100 nM bufalin for 48 h. SGC7901 cells were
treated with 5 µM GDC0068 and/or 10 µM Fumonisin B1
for 6 h, then were treated with 0.10 µM cisplatin and/or
0.100 nM bufalin under normoxic or hypoxic conditions for 48 h and
were then harvested. Cell viability was measured with a Cell
Counting kit-8 (CCK-8) kit (Dojindo Molecular Technologies, Inc.,
Gaithersburg, MD, USA), followed by the addition of 10 µl
CCK-8 solution. The cells were then incubated for 2 h at 37°C, then
optical density was read at 450 nm. All experiments were performed
in triplicate. Apoptosis was analyzed by flow cytometry as
previously described (21,27). In brief, cells were seeded at a
density of 15×105 cells/well in 6-well plates, cultured
with different reagents for 48 h and harvested. Cells were washed
with phosphate-buffered saline and then stained with 5 µl
Annexin V and 5 µl propidium iodide for 15 min in the dark
at room temperature according to the manufacturers instructions (BD
Biosciences, San Jose, CA, USA). The apoptotic rate (%) of the
stained cells was analyzed using an Epics Altra II flow cytometer
(Beckman Coulter, Inc., Brea, CA, USA). Experiments were repeated
three times.
Immunoblotting
Protein concentrations of cell lysates were
determined using the Bio-Rad protein assay (Bio-Rad, Richmond, CA,
USA). Lysates were resolved by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis; proteins were
transferred to membranes and immunoblotted as previously described
(21,27) In brief, cells were treated with
different reagents for 48 h, then lysed in lysis buffer [2% sodium
dodecyl sulfate (SDS), 10% glycerol, 62.5 mM Tris-HCl, pH 6.8].
Equal amounts of cell lysate (20 µg) were resolved on
SDS-polyacrylamide gels, and the proteins were transferred to a
nitrocellulose membrane. The membrane was blocked with 10% non-fat
milk in Tris-buffered saline containing 0.1% (v/v) Tween-20 for 50
min and then incubated again with 10% non-fat milk containing the
primary antibodies for 2 h at room temperature. Subsequent to
washing, the membrane was incubated with 10% non-fat milk
containing the secondary antibodies conjugated with horseradish
peroxidase for 1 h at room temperature. The proteins were
visualized with 5-bromo-4-chloro-3-indolyl phosphate/nitro blue
tetrazolium (Tiangen Biotech Co., Ltd., Beijing, China). Blots were
stained with the anti-β-actin antibody, the relative level (%) of
each protein was normalized to the β-actin band density, and were
evaluated using ImageJ software, version 1.46 (National Institutes
of Health, Bethesda, MD, USA).
Statistical analysis
All data are expressed as the mean ± standard
deviation. Comparisons were made using one-way analysis of variance
followed by Dunnett's t-test. Statistical comparisons were
performed using SPSS software, version 13.0 (SPSS, Inc., Chicago,
IL, USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
Cisplatin- and hypoxia-induced AKT
activation contributes to cisplatin resistance in GC cells
The effects of cisplatin and/or hypoxia on AKT
expression and activation in GC cells were investigated. As
predicted, SGC7901, MKN-45 and BGC823 cells treated with cisplatin
or cultured under hypoxia had increased p-AKT levels when compared
with cells in normoxic conditions. When cells were treated with
cisplatin under hypoxia, the p-AKT levels were increased compared
with that of cells treated with cisplatin or hypoxia alone
(Fig. 1A). Levels of total AKT
were not significantly altered by either cisplatin or hypoxia in
the three cell lines (Fig.
1A).
To investigate the exact role of AKT activation in
cisplatin resistance, GDC0068 (a specific AKT inhibitor) and
Fumonisin B1 (a specific AKT activator) was introduced to SGC7901
cells. The antiproliferation effect of cisplatin was significantly
facilitated by GDC0068 and attenuated by Fumonisin B1 in SGC7901
cells, under normoxic and hypoxic conditions (Fig. 1B). Consistent with these results,
cisplatin-induced apoptosis rates were also significantly increased
by GDC0068 and attenuated by Fumonisin B1 in SGC7901 cells, in
normoxic and hypoxic conditions (Fig.
1C and D). In addition, it was demonstrated that the efficiency
of cisplatin in SGC7901 cells was significantly limited by hypoxia
(Fig. 1B–D).
Bufalin synergizes with cisplatin to
inhibit growth and induce apoptosis in GC cells
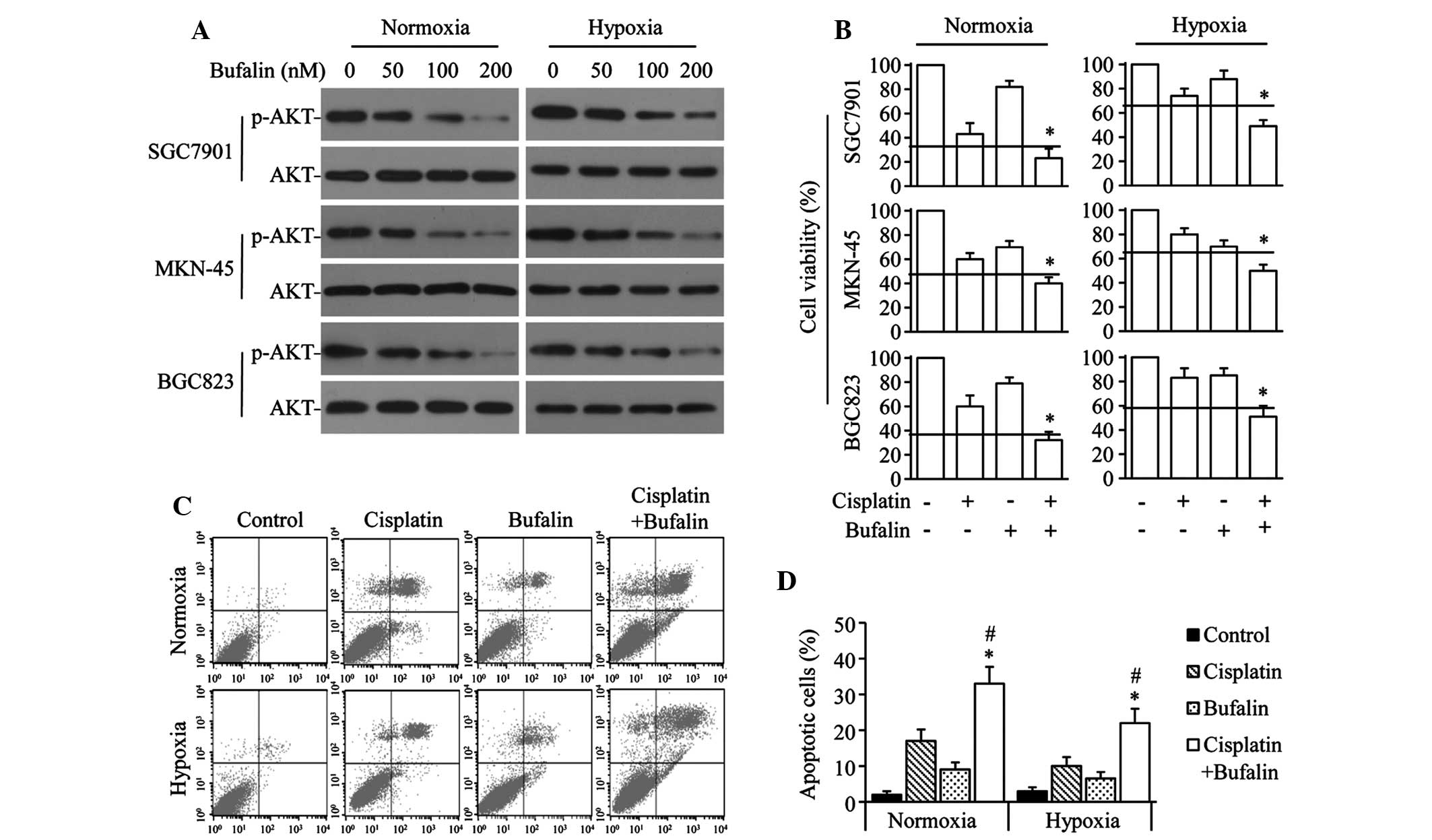
To investigate the effect of bufalin on AKT
activation in GC cells, SGC7901, MKN-45 and BGC823 cells were
exposed to varying doses of bufalin under normoxic or hypoxic
conditions. Bufalin significantly downregulated p-AKT levels in a
dose-dependent manner, but did not significantly alter AKT levels
under either normoxia or hypoxia (Fig.
2A). The combined effect of bufalin and cisplatin was also
investigated in GC cells. Bufalin synergistically enhanced the
antiproliferation effect of cisplatin in normoxic or hypoxic
conditions in the 3 GC cell lines (Fig. 2B). Similarly, cisplatin+bufalin
significantly increased the number of apoptotic cells compared with
the effect of cisplatin or bufalin alone under either normoxic or
hypoxic conditions in SGC7901 cells (Fig. 2C and D).
Bufalin eradicates cisplatin-induced
activation of the AKT pathway in SGC7901 cells
The mechanism underlying the anticancer effect of
cisplatin+bufalin in SGC7901 cells was investigated. Cells treated
with cisplatin exhibited markedly increased expression of p-AKT and
its downstream molecules p-GSK3β, p-mTOR, P-4EBP1 and p-S6K;
whereas cells treated with bufalin exhibited significantly reduced
expression of these five proteins in normoxic and hypoxic
conditions, compared with untreated cells (Fig. 3).
Cisplatin-resistant GC cells show
sensitivity to bufalin with active AKT pathways
To examine the contribution of the AKT pathway to
acquired cisplatin resistance, SGC7901 cells were exposed to
gradually increasing concentrations of cisplatin. After 6 months of
culture, the resulting SGC7901-CR cells had recovered their
proliferative capacity comparable to that of parental cells
(Fig. 4A). However, the acquired
resistance of SGC7901-CR cells was reversed by bufalin (Fig. 4B). Flow cytometry showed that
bufalin induced a significant increase in the number of apoptotic
SGC7901-CR cells compared with untreated controls (Fig. 4C and D). Although SGC7901-CR cells
expressed higher levels of p-AKT, p-GSK3β, p-mTOR, p-4EBP1 and
p-S6K than their parental SGC7901 cells (Fig. 4E), SGC7901-CR cells treated with
bufalin expressed significantly less of these five proteins than
untreated SGC7901-CR cells (Fig.
4E).
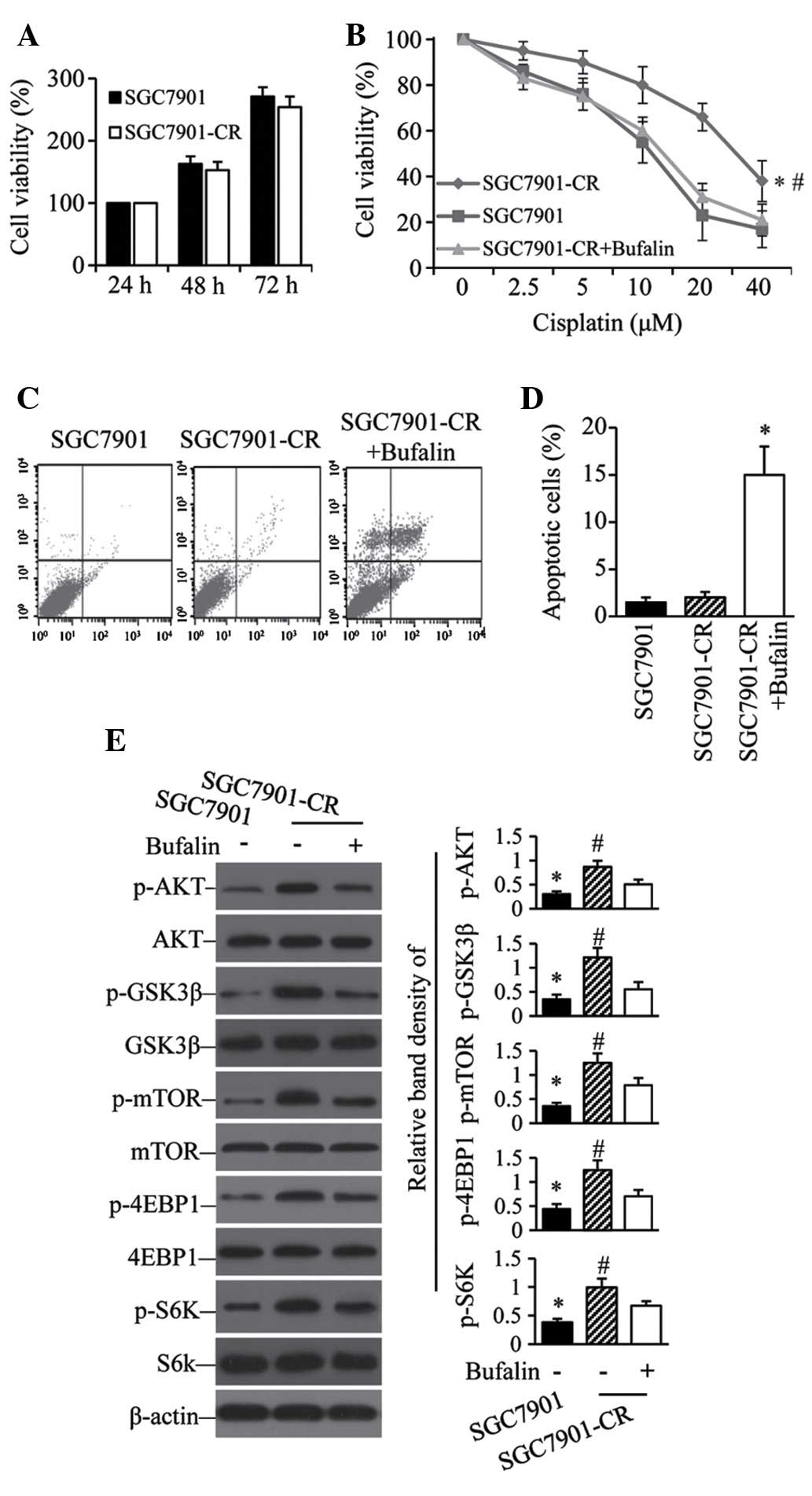 | Figure 4Activation of the AKT pathway
contributes to acquired cis-platin resistance. (A) SGC7901 and
SGC7901-CR cells were cultured for 24, 48 or 72 h; cell viability
was then measured. (B) SGC7901 cells and SGC7901-CR cells treated
without or with bufalin (100 nM) were cultured with indicated
concentrations of cisplatin for 48 h; cell viability was then
measured. *P<0.05 compared with SGC7901 cells.
#P<0.05 compared with SGC7901CR + bufalin cells.
SGC7901 cells and SGC7901-CR cells treated without or with bufalin
(100 nM) were cultured for 48 h. Cells were subjected to flow
cytometry to analyze apoptosis. (C) Representative dot plots are
shown. (D) Apoptosis rates *P<0.05 compared with
SGC7901-CR cells. (E) Cell lysates were immunoblotted and the
density of each band was measured. Band densities of p-AKT,
p-GSK3β, p-mTOR, P-4EBP1 and p-S6K were normalized to the
respective total form. *P<0.05 compared with
untreated cells. #P<0.05 compared with cells treated
with cisplatin. CR, cisplatin resistant; p-, phosphorylated; GSK,
glycogen synthase; mTOR, mammalian target of rapamycin; 4EBP1,
eukaryotic translation initiation factor 4E binding protein 1; S6K,
ribosomal protein S6 kinase. |
Discussion
Cisplatin remains a common chemotherapeutic agent
for GC (3). Unfortunately,
intrinsic and acquired resistance to cisplatin is common. The
present study demonstrated that activation of the AKT pathway
contributes to intrinsic and acquired resistance to cisplatin, and
blocking the AKT pathway with bufalin enhances the efficacy of
cisplatin in GC cells.
Bufalin, a bioactive component of skin and parotid
venom glands of the Venenum bufonis toad (20), has been tested in clinical trials
to treat advanced hepatocellular carcinoma, non-small-cell lung
cancer and pancreatic cancer (28); and has been shown experimentally to
inhibit cancer through the AKT pathway (22–25).
In the present study, bufalin inhibited the activation of AKT and
its downstream molecules GSK-3β, mTOR, S6K and 4EBP1; and when
combined with cisplatin, inhibited proliferation and promoted
apoptosis of GC cells by diminishing activation of the AKT y under
normoxic and hypoxic conditions. Bufalin also reversed acquired
cisplatin resistance and induced apoptosis in SGC7901-CR cells
through the AKT pathway.
AKT is a member of a family of serine/threonine
kinases and acts downstream of PI3K to phosphorylate various
molecules involved in several cancer-related processes (7,8,29).
Activation of the ATK pathway is frequently implicated in
resistance to anticancer therapies; thus, inhibitors of the AKT
pathway are being evaluated in pre-clinical and clinical studies to
determine whether they can restore sensitivity to combinations of
therapeutic agents (7,30–32).
Overexpression and activation of AKT has been shown to contribute
to cisplatin resistance in GC cells (10–13).
Cisplatin is generally considered a cytotoxic drug that kills
cancer cells by damaging DNA, inhibiting DNA synthesis and mitosis,
and inducing apoptosis (33).
Cisplatin-induced DNA damage reportedly triggers PI3K/AKT
activation, which in turn phosphorylates its downstream molecules
and promotes survival of cancer cells (34,35).
The present study demonstrated that AKT activation causes intrinsic
and acquired resistance to cisplatin in GC cells. p-AKT upregulates
its downstream molecules p-GSK3β, p-mTOR, P-4EBP1 and p-S6K.
Treatment with a combination of bufalin and cisplatin inhibited
p-AKT, and inhibited the growth and induced apoptosis of GC
cells.
Another intrinsic cause of cisplatin resistance is
the hypoxic microenvironment that commonly exists in solid tumors
(13,14,17).
The aberrant microvasculature of solid tumors leads to deficient
oxygen delivery in certain regions, creating zones of hypoxia
(16). Thus, attacking normoxic
and hypoxic cells, which are mixed within the tumors in different
regions of differing oxygen partial pressures, is a reasonable
strategy to treat solid cancers. The present study shows that
hypoxic GC cells were refractory to cisplatin-induced growth
inhibition and apoptosis and had high levels of p-AKT and its
downstream molecules p-GSK3β, p-mTOR, P-4EBP1 and p-S6K.
Downregulation of p-AKT by treatment with bufalin significantly
improved the efficacy of cisplatin in hypoxic GC cells. These data
show that hypoxia induces cisplatin resistance at least partially
by activating the AKT pathway. Hypoxia-induced AKT activation has
been elucidated in a number of types of cancer (36). Reportedly, AKT activation increases
expression of HIF-1α (13,18,19),
which in turn aggravates hypoxia. This indicates positive feedback,
whereby hypoxia is induced by p-AKT and HIF-1α, which increases
cisplatin resistance. Thus breaking the cycle by inhibiting AKT is
a reasonable strategy to overcome cisplatin resistance under
hypoxia.
In conclusion, the present results indicate that
administering cisplatin in combination with bufalin may overcome or
delay resistance to cisplatin and extend the efficacy of cisplatin
in treating GC, thus suggesting a novel strategy for treatment of
advanced GC to be investigated further in clinical trials.
Acknowledgments
This study was supported by Health Department of
Heilongjiang Province of China (grant no. 2014–013).
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
2
|
Shen L, Shan YS, Hu HM, Price TJ, Sirohi
B, Yeh KH, Yang YH, Sano T, Yang HK, Zhang X, et al: Management of
gastric cancer in Asia: Resource-stratified guidelines. Lancet
Oncol. 14:e535–e547. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pasini F, Fraccon AP and DE Manzoni G: The
role of chemotherapy in metastatic gastric cancer. Anticancer Res.
31:3543–3554. 2011.PubMed/NCBI
|
|
4
|
Matt P, van Zwieten-Boot B, Calvo Rojas G,
Ter Hofstede H, Garcia-Carbonero R, Camarero J, Abadie E and
Pignatti F: The European medicines agency review of
Tegafur/Gimeracil/Oteracil (TeysunoT™) for the treatment of
advanced gastric cancer when given in combination with cisplatin:
Summary of the scientific assessment of the committee for medicinal
products for human use (CHMP). Oncologist. 16:1451–1457. 2011.
View Article : Google Scholar
|
|
5
|
Okuno T, Shirotsuki J, Murahashi K and
Sawada T: Advanced gastric cancer (stage IV) leading to perforation
during chemotherapy with S-1 plus cisplatin. Gan To Kagaku Ryoho.
41:1313–1315. 2014.In Japanese. PubMed/NCBI
|
|
6
|
Sasaki T and Kuniyasu H: Significance of
AKT in gastric cancer (Review). Int J Oncol. 45:2187–2192.
2014.PubMed/NCBI
|
|
7
|
Polivka J Jr and Janku F: Molecular
targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol
Ther. 142:164–175. 2014. View Article : Google Scholar
|
|
8
|
Radisavljevic Z: AKT as locus of cancer
multidrug resistance and fragility. J Cell Physiol. 228:671–674.
2013. View Article : Google Scholar
|
|
9
|
Nam SY, Lee HS, Jung GA, Choi J, Cho SJ,
Kim MK, Kim WH and Lee BL: Akt/PKB activation in gastric carcinomas
correlates with clinicopathologic variables and prognosis. APMIS.
111:1105–1113. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang LL, Zhang J, Shen L, Xu XM and Yu
HG: Overexpression of AKT decreases the chemosensitivity of gastric
cancer cells to cisplatin in vitro and in vivo. Mol Med Rep.
7:1387–1390. 2013.PubMed/NCBI
|
|
11
|
Zhang J, Zhang LL, Shen L, Xu XM and Yu
HG: Regulation of AKT gene expression by cisplatin. Oncol Lett.
5:756–760. 2013.PubMed/NCBI
|
|
12
|
Park J, Ko YS, Yoon J, Kim MA, Park JW,
Kim WH, Choi Y, Kim JH, Cheon Y and Lee BL: The forkhead
transcription factor FOXO1 mediates cisplatin resistance in gastric
cancer cells by activating phosphoinositide 3-kinase/Akt pathway.
Gastric Cancer. 17:423–430. 2014. View Article : Google Scholar
|
|
13
|
Sun XP, Dong X, Lin L, Jiang X, Wei Z,
Zhai B, Sun B, Zhang Q, Wang X, Jiang H, et al: Up-regulation of
survivin by AKT and hypoxia-inducible factor 1α contributes to
cisplatin resistance in gastric cancer. FEBS J. 281:115–128. 2014.
View Article : Google Scholar
|
|
14
|
Rohwer N, Dame C, Haugstetter A,
Wiedenmann B, Detjen K, Schmitt CA and Cramer T: Hypoxia-inducible
factor 1alpha determines gastric cancer chemosensitivity via
modulation of p53 and NF-kappaB. PLoS One. 5:e120382010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Adamski J, Price A, Dive C and Makin G:
Hypoxia-induced cytotoxic drug resistance in osteosarcoma is
independent of HIF-1alpha. PLoS One. 8:e653042013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wilson WR and Hay MP: Targeting hypoxia in
cancer therapy. Nat Rev Cancer. 11:393–410. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rankin EB and Giaccia AJ: The role of
hypoxia-inducible factors in tumorigenesis. Cell Death Differ.
15:678–685. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee SH, Jee JG, Bae JS, Liu KH and Lee YM:
A group of novel hif-1alpha inhibitors, glyceollins, blocks
HIF-1alpha synthesis and decreases its stability via inhibition of
the PI3K/AKT/mTOR pathway and Hsp90 binding. J Cell Physiol.
230:853–862. 2014. View Article : Google Scholar
|
|
19
|
Kim BR, Yoon K, Byun HJ, Seo SH, Lee SH
and Rho SB: The anti-tumor activator sMEK1 and paclitaxel
additively decrease expression of HIF-1α and VEGF via
mTORC1-S6K/4E-BP-dependent signaling pathways. Oncotarget.
5:6540–6551. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang Z, Luo H, Wang H and Hou H:
Preparative isolation of bufalin and cinobufagin from Chinese
traditional medicine Chansu. J Chromatogr Sci. 46:81–85. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hu F, Han J, Zhai B, Ming X, Zhuang L, Liu
Y, Pan S and Liu T: Blocking autophagy enhances the apoptosis
effect of bufalin on human hepatocellular carcinoma cells through
endoplasmic reticulum stress and JNK activation. Apoptosis.
19:210–223. 2014. View Article : Google Scholar
|
|
22
|
Zhang ZJ, Yang YK and Wu WZ: Bufalin
attenuates the stage and metastatic potential of hepatocellular
carcinoma in nude mice. J Transl Med. 12:572014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhu Z, Sun H, Ma G, Wang Z, Li E and Liu Y
and Liu Y: Bufalin induces lung cancer cell apoptosis via the
inhibition of PI3K/Akt pathway. Int J Mol Sci. 13:2025–2035. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li M, Yu X, Guo H, Sun L, Wang A, Liu Q,
Wang X and Li J: Bufalin exerts antitumor effects by inducing cell
cycle arrest and triggering apoptosis in pancreatic cancer cells.
Tumour Biol. 35:2461–2471. 2014. View Article : Google Scholar
|
|
25
|
Tsai SC, Lu CC, Lee CY, Lin YC, Chung JG,
Kuo SC, Amagaya S, Chen FN, Chen MY and Chan SF: AKT
serine/threonine protein kinase modulates bufalin-triggered
intrinsic pathway of apoptosis in CAL 27 human oral cancer cells.
Int J Oncol. 41:1683–1692. 2012.PubMed/NCBI
|
|
26
|
Zhai B, Hu F, Jiang X, Xu J, Zhao D, Liu
B, Pan S, Dong X, Tan G, Wei Z, et al: Inhibition of Akt reverses
the acquired resistance to sorafenib by switching protective
autophagy to autophagic cell death in hepatocellular carcinoma. Mol
Cancer Ther. 13:1589–1598. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao D, Zhai B, He C, Tan G, Jiang X, Pan
S, Dong X, Wei Z, Ma L, Qiao H, et al: Upregulation of HIF-2α
induced by sorafenib contributes to the resistance by activating
the TGF-α/EGFR pathway in hepatocellular carcinoma cells. Cell
Signal. 26:1030–1039. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Meng Z, Yang P, Shen Y, Bei W, Zhang Y, Ge
Y, Newman RA, Cohen L, Liu L, Thornton B, et al: Pilot study of
huachansu in patients with hepatocellular carcinoma, nonsmall-cell
lung cancer, or pancreatic cancer. Cancer. 115:5309–5318. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dillon RL and Muller WJ: Distinct
biological roles for the akt family in mammary tumor progression.
Cancer Res. 70:4260–4264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Burris HA III: Overcoming acquired
resistance to anticancer therapy: Focus on the PI3K/AKT/mTOR
pathway. Cancer Chemother Pharmacol. 71:829–842. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Molife LR, Yan L, Vitfell-Rasmussen J,
Zernhelt AM, Sullivan DM, Cassier PA, Chen E, Biondo A, Tetteh E,
Siu LL, et al: Phase 1 trial of the oral AKT inhibitor MK-2206 plus
carboplatin/paclitaxel, docetaxel, or erlotinib in patients with
advanced solid tumors. J Hematol Oncol. 7:12014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Davies BR, Greenwood H, Dudley P, Crafter
C, Yu DH, Zhang J, Li J, Gao B, Ji Q, Maynard J, et al: Preclinical
pharmacology of AZD5363, an inhibitor of AKT: Pharmacodynamics,
antitumor activity and correlation of monotherapy activity with
genetic background. Mol Cancer Ther. 11:873–887. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stronach EA, Chen M, Maginn EN, Agarwal R,
Mills GB, Wasan H and Gabra H: DNA-PK mediates AKT activation and
apoptosis inhibition in clinically acquired platinum resistance.
Neoplasia. 13:1069–1080. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li M, Balch C, Montgomery JS, Jeong M,
Chung JH, Yan P, Huang TH, Kim S and Nephew KP: Nephew, Integrated
analysis of DNA methylation and gene expression reveals specific
signaling pathways associated with platinum resistance in ovarian
cancer. BMC Med Genomics. 2:342009. View Article : Google Scholar
|
|
36
|
Stegeman H, Kaanders JH, Wheeler DL, van
der Kogel AJ, Verheijen MM, Waaijer SJ, Iida M, Grénman R, Span PN
and Bussink J: Activation of AKT by hypoxia: A potential target for
hypoxic tumors of the head and neck. BMC Cancer. 12:4632012.
View Article : Google Scholar : PubMed/NCBI
|