Introduction
Congenital stationary night blindness (CSNB) is a
group of disorders characterized by non-progressive retinal
dysfunction (1). CSNB is caused by
mutations in the phototransduction component or mutations in
retinal signaling from the outer retina to the inner retina
(1). There are two main types of
CSNB with specific fundus findings, fundus albipunctatus and Oguchi
disease. These disorders are inherited in an autosomal recessive
pattern (2,3). A golden-brown or diffuse gray-white
fundus discoloration is observed in Oguchi disease in the light
adapted state. Following complete dark adaptation, the fundus
returns to normal (1,2). Oguchi disease is caused by the
mutations in G-protein-dependent receptor kinase 1 (GRK1)
(MIM:180381) and S-antigen (SAG) (MIM:181031) (4). Patients are divided into two groups
type 1 or type 2 based on mutations in two genes, namely,
SAG and GRK1, respectively (2).
The aim of this study was to investigate the
presence of these mutations, yet to be described in a Turkish
population, in family members with newly diagnosed Oguchi
disease.
Materials and methods
Family history
Four family members with night blindness were
referred to the Ulucanlar Eye Hospital Retina Clinic (Ankara,
Turkey). Four siblings: A 12-year-old male (case 1), 14-year-old
female (case 2), 16-year-old female (case 3), 19-year-old female
(case 4), and their 41-year-old mother (case 5) and 44-year-old
father (case 6) were examined. Cases 1–4 suffered night blindness
since early childhood. The mother and father were 4th
degree relatives (first cousins) and thus had a consanguineous
marriage. None of the cases had diabetes, hypertension, a history
of systemic or ocular disease, nor had any undergone any
surgery.
A complete ophthalmological examination was
conducted including visual acuity, intraocular pressure measurement
with Goldmann applanation tonometry, biomicroscobic examination and
dilated pupil examination of the posterior segment.
Ethical approval
Study procedures were conducted in accordance with
the Declaration of Helsinki. The study protocol was approved by The
Ethical Committee of Diskapi Training and Research Hospital
(Ankara, Turkey) and written informed content was obtained from all
study participants. All patients were Turkish Caucasians. This
study is registered as an Australian New Zealand Clinical Trials
Registry (no. ACTRN 368991).
Mutation testing and Sanger
sequencing
All six family members underwent mutation testing.
Whole blood (10 ml) was taken from each family member. Genomic DNA
was obtained from peripheral leukocytes by ammonium acetate
extraction (AppliChem GmbH, Gatersleben, Germany). To conduct
Sanger sequencing, DNA samples were quantified with a NanoDrop 2000
spectrophotometer (Thermo Fisher Scientific, Inc., Wilmington, NC,
USA) prior to polymerase chain reaction (PCR). Primer sequences
(Table I) used for PCR reactions
were determined manually by using Ensembl Database ID's
ENST00000335678 (GRK1) and ENST00000409110 (SAG).
Then 200 ng genomic DNA was combined with 5X Buffer (Promega
Corporation, Madison, WI, USA), 0.2 mM of each dNTP (Promega
Corporation), 1.5 mM MgCl2 (Promega Corporation), 0.2
µM of forward and reverse primers (IDT, Inc., Coralville,
IA, USA), 1 unit GoTaq DNA Polymerase (Promega Corporation) and
nuclease-free water up to 25 µl. The PCR mix was vortex
mixed, centrifuged at 650 × g for 1 min, then transferred to a
thermal cycler (ABI 9700; Applied Biosystems, Foster City, CA,
USA). Samples were amplified with an initial heat denaturation step
of 94°C for 5 min; followed by 35 cycles of 94°C for 30 sec,
54–65°C for 30 sec, and 72°C for 30 sec, and a final of extension
at 72°C for 7 min. Primer sequences and annealing temperatures are
shown in Table I. PCR products
were run on agarose gels and samples with the correct band size
were purified using Wizard SV Gel and PCR Cleanup system (Promega
Corporation, Madison, WI, USA). After purification, PCR products
were cycle sequenced using forward primers and the BigDye
Terminator v3.1 Cycle Sequencing kit (Thermo Fisher Scientific,
Inc.). After cycle sequencing, products were cleaned up using ZR
DNA Sequencing Clean up kit (Zymo Research, Irvine, CA, USA) and
loaded onto 96-well plates of a 3130 Genetic Analyzer (Applied
Biosystems). Analysis of the results was conducted using SeqScape 3
Software (Applied Biosystems).
 | Table IPrimer details for polymerase chain
reaction. |
Table I
Primer details for polymerase chain
reaction.
A, SAG primer
details: ENSEMBL: ENST00000409110
|
|---|
| Exon | Amplicon size
(bp) | Annealing temperature
(°C) | Primer sequences |
|---|
| Exon2 | 276 | 54 | SAG-E2F:
5′-GGATCTCGTGAGTAGGTTTC-3′ |
| | | SAG-E2R:
5′-CACTGTACTTGAAAAAGCTCC-3′ |
| Exon3 | 294 | 60 | SAG-E3F:
5′-CATATTGGCCAGGCTCAAAC-3′ |
| | | SAG-E3R:
5′-AAAGTGAGCGGTTATCTGTGAC-3′ |
| Exon4 | 330 | 54 | SAG-E4F:
5′-AATGAACATGGATTACATGTG-3′ |
| | | SAG-E4R:
5′-GTCACGTGAATTAGGTACAGG-3′ |
| Exon5 | 342 | 63 | SAG-E5F:
5′-GGTTGAAAACCCGTGTTCGC-3′ |
| | | SAG-E5R:
5′-CACATGATAAGGTGCTGCGG-3′ |
| Exon6 | 218 | 60 | SAG-E6F:
5′-TAATGGAACAGCCCCTTCTG-3′ |
| | | SAG-E6R:
5′-CCCAGCATTGGTGACAGAGT-3′ |
| Exon7 | 319 | 60 | SAG-E7F:
5′-TGCAACCCCGAATAGGACAT-3′ |
| | | SAG-E7R:
5′-CAGCCCTATGGGAAGAGGTCT-3′ |
| Exon8 | 359 | 60 | SAG-E8F:
5′-GAGCATTCCTGGAGAATCTCC-3′ |
| | | SAG-E8R:
5′-GAAACAAGCTTCCTTGCAAGG-3′ |
| Exon9 | 336 | 60 | SAG-E9F:
5′-GTGTTTCAGGCCCTTCCTTAG-3′ |
| | | SAG-E9R:
5′-CAGACCAGAGAAGTGACCTCTC-3′ |
| Exon10 | 431 | 60 | SAG-E10F:
5′-ACAGGACTTCAAAACCCCAG-3′ |
| | | SAG-E10R:
5′-GGTGTGGTAGATGCAGAGCTAAG-3′ |
| Exon11 | 360 | 60 | SAG-E11F:
5′-GTCAAGTTCCCAGGCTCTTG-3′ |
| | | SAG-E11R:
5′-CAGGGTGATGTGAAGGGAAG-3′ |
| Exon12 | 226 | 60 | SAG-E12F:
5′-CTGCCCATCTGCTCTTCACC-3′ |
| | | SAG-E12R:
5′-CTCCCAGTCATTCAGGAAAGG-3′ |
| Exon13 | 252 | 60 | SAG-E13F:
5′-GATGTTGTGAGTTCGGGTGC-3′ |
| | | SAG-E13R:
5′-CACAACTGTCCAGAAAGCAGC-3′ |
| Exon14 | 248 | 63 | SAG-E14F:
5′-TGTGACTCTCCGCAGCCATAG-3′ |
| | | SAG-E14R:
5′-CACTCCCATGCTCTGAGATGC-3′ |
| Exon15 | 251 | 60 | SAG-E15F:
5′-ACGCAGTGATCATGAACTGC-3′ |
| | | SAG-E15R:
5′-GACTCAAAGAGGGTTTTGTGC-3′ |
| Exon16 | 348 | 63 | SAG-E16F:
5′-CCTTGATCAGTTCCTTCGTTGC-3′ |
| | | SAG-E16R:
5′-CCAGGGGAGAACAAACAAGCT-3′ |
B, GRK1 primer
details: ENSEMBL: ENST00000335678
|
|---|
| Exon | Amplicon size
(bp) | Annealing temperature
(°C) | Primer sequences |
|---|
| Exon1/1 | 443 | 64 | GRK1-E1/1F:
5′-TGCTCTGTCTGTGAACGCTCC-3′ |
| | | GRK1-E1/1R:
5′-AGAAGAGTTTGGCCTGGGGG-3′ |
| Exon1/2 | 526 | 64 | GRK1-E1/2F:
5′-CGGCAGACAATGACCTCCAG-3′ |
| | | GRK1-E1/2R:
5′-AGGCACCAGCTGTTAAGGGC-3′ |
| Exon2 | 280 | 62 | GRK1-E2F:
5′-CGATGCACCTAGTCCCTTTCC-3′ |
| | | GRK1-E2R:
5′-ATGGCTCTGCCTGTGGAAAG-3′ |
| Exon3 | 360 | 62 | GRK1-E3F:
5′-TCAAAACGACCAGAACGCTG-3′ |
| | | GRK1-E3R:
5′-TCGTGAGGTTGTGCAGAGACC-3′ |
| Exon4 | 207 | 64 | GRK1-E4F:
5′-TGTGCAGCCAGGGGTGACTC-3′ |
| | | GRK1-E4R:
5′-GTATGTGCAAGTGCACACAGGC-3′ |
| Exon5 | 291 | 62 | GRK1-E5F:
5′-AGCATCAGTCCTGCGATTCC-3′ |
| | | GRK1-E5R:
5′-CAGTAACGATCCCATCACTGCC-3′ |
| Exon6 | 353 | 62 | GRK1-E6F:
5′-TCTGGTCTGACCACCCAAGAG-3′ |
| | | GRK1-E6R:
5′-CCGACTCTCACAGGCTGGAC-3′ |
| Exon7 | 451 | 64 | GRK1-E7F:
5′-GGCTAAACGGCGCTTCCTTC-3′ |
Bioinformatics analysis
Two of the most popular bioinformatics tools
[Sorting Intolerant from Tolerant (SIFT; http://sift.bii.a-star.edu.sg/) and Polymorphism
Phenotyping v2 (PolyPhen-2; http://genetics.bwh.harvard.edu/pph2/)] were used to
predict whether the amino acid substitutions identified in the
study affect protein function. These types of tools use predictions
based on the degree of conservation of amino acid residues in
sequence alignments derived from closely associated sequences, and
additionally the impact of the substitution on the structure and
function of the protein.
Results
Patient characteristics
The family members (four siblings 12-year-old male,
14-year-old female, 16-year-old female and 19-year-old, and their
41-year-old mother and a 44-year-old father were examined. There
was no history of night blindness in either the mother or the
father. However, four of the family members had a history of
nyctalopia since early childhood. Their visual acuity was 20/20
bilaterally and they had no refractive error. All children and
their parents had normal intraocular eye pressure (11–19 mmHg)
values. All patient's eyes were phakic and slit-lamp examination
revealed normal findings. Fundus examination of parents revealed
normal findings. However, fundus images of the four children showed
a characteristic golden metallic reflex in all areas of the fundus.
The Mizuo-Nakamura phenomenon was demonstrated: The fundus color
changed to normal after three hours of dark adaptation.
Sanger sequencing results
Sanger sequencing for two candidate genes,
GRK1 and SAG was conducted. The coding regions and
their flanking sequences were analyzed in the 4 siblings and their
parents to identify mutations that may be causing Oguchi disease.
Sequencing results were analyzed with SeqScape 3 software (Applied
Biosystems) by aligning to the Human Reference Genome Sequence
(GRCh38/hg38), and single nucleotide variations (SNVs) were
determined. The single nucleotide polymorphism (SNP) database dbSNP
(build 141) served as a reference for registered SNPs, and
non-synonymous SNVs were extracted. Heterozygous and homozygous
variations were identified, including de novo heterozygous
variations and a homozygous variation, as presented in Table II. Molecular analysis of the
SAG gene revealed no SNPs that segregated with the disease.
The mutation analysis of the other candidate gene, GRK1,
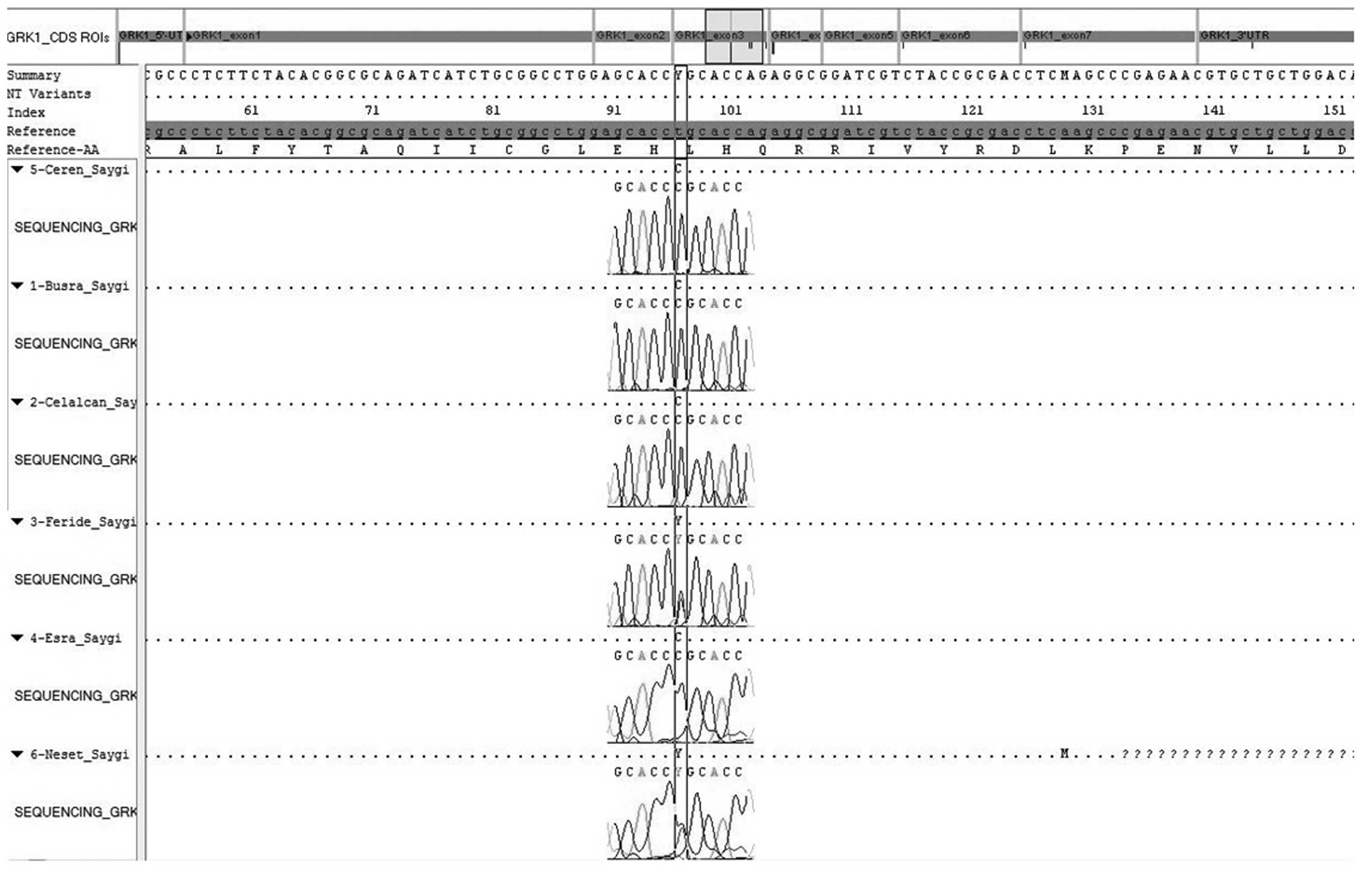
demonstrated a novel mutation (c.923T>C) (Fig. 1), shared by all family members,
which was homozygous in all siblings and heterozygous in the two
parents, consistent with autosomal recessive transmission (Table II). This novel finding directed us
to determine whether this position is conserved phylogenetically
among other GRK1 orthologs by utilizing Clustal W2.1 (Conway
Institute UCD, Dublin, Ireland). Nucleotide changes resulting in
the change of the strictly conserved residue 308 from leucine to
proline affects the protein kinase catalytic domain of the gene.
Popular bioinformatics tools SIFT and PolyPhen-2 analysis scores (0
and 1.000, respectively) confirm this change to be damaging.
Further analysis is required in healthy populations to confirm
pathogenicity. No other mutations were found in the GRK1
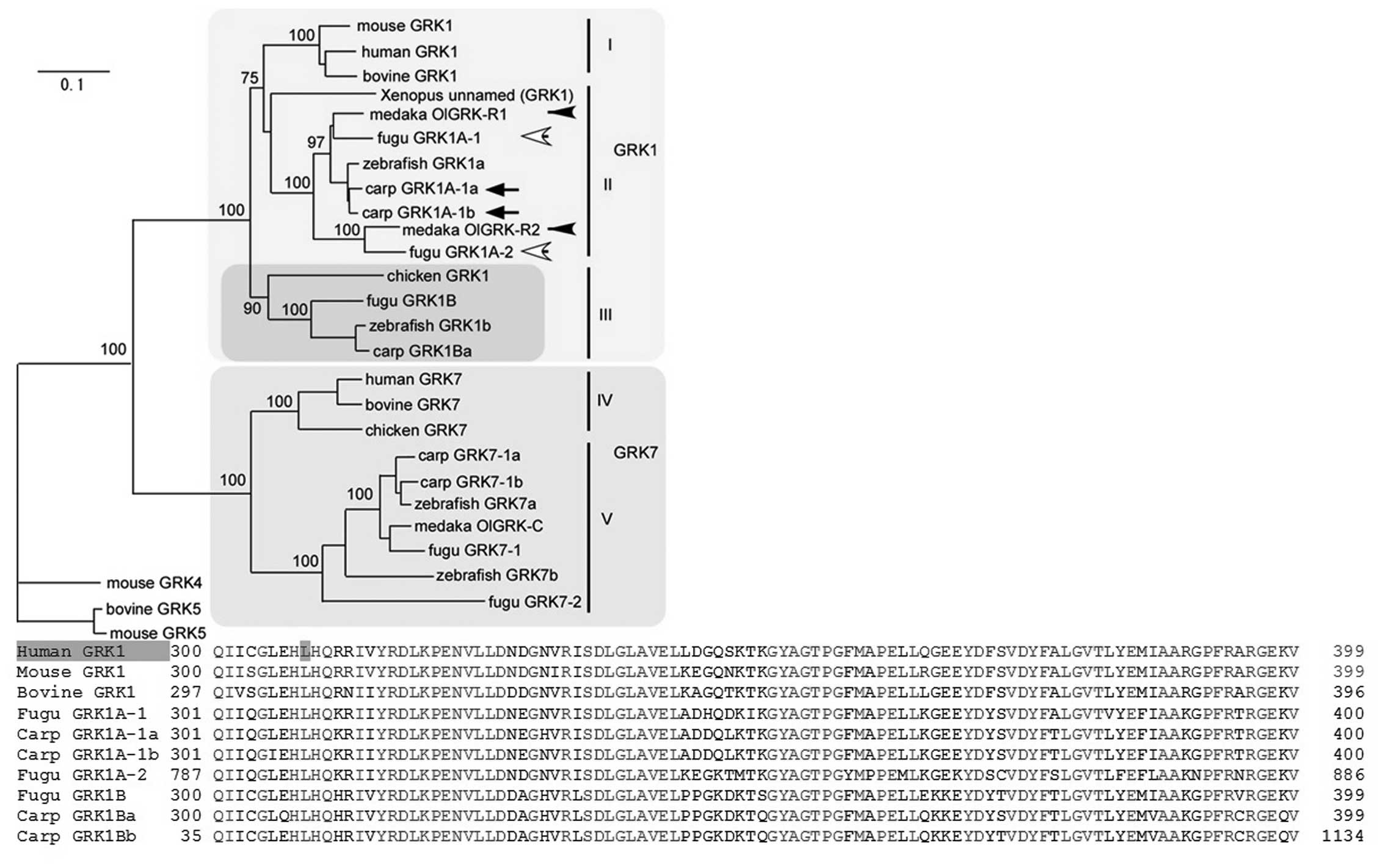
gene in the patients. The molecular phylogenetic tree of vertebrate
GRK1 genes, which are highly conserved, is shown in Fig. 2. Branch length is proportional to
the number of amino acid substitutions; the scale bar represents
0.1 amino acid substitutions per position in the sequences. As the
branches are short, we can therefore predict damaging effects from
the occurrence of amino acid changes (5).
| Table IIAmino acid alterations in SAG
and GRK1 genes. |
Table II
Amino acid alterations in SAG
and GRK1 genes.
A, SAG amino acid
alterations
|
|---|
| Case | Gene region | Alteration | Condition | SNP ID | Amino acid
alterations |
|---|
| 1 | Exon 16 | c.1207G>A | Heterozygote | rs1046974 | Val403Ile |
| 2 | Exon 16 | c.1207G>A | Homozygote | rs1046974 | Val403Ile |
| Intron 4–5 | c.181+82A>G | Homozygote | rs2304777 | – |
| Intron 6–7 | c.436−18G>C | Homozygote | rs2304774 | – |
| Intron 9–10 | c.733+31T>G | Homozygote | rs745498 | – |
| 3 | Exon 16 | c.1207G>A | Homozygote | rs1046974 | Val403Ile |
| 4 | Exon 16 | c.1207G>A | Heterozygote | rs1046974 | Val403Ile |
| 5 | Exon 16 | c.1207G>A | Homozygote | rs1046974 | Val403Ile |
| 6 | Exon 16 | c.1207G>A | Heterozygote | rs1046974 | Val403Ile |
B, GRK1 amino acid
alterations
|
|---|
| Case | Gene region | Alteration | Condition | SNP ID | Amino acid
alterations |
|---|
| 1 | Exon 3 | c.923T>C | Homozygote | Novel | Leu308Pro |
| 2 | Exon 3 | c.923T>C | Homozygote | Novel | Leu308Pro |
| 3 | Exon 3 | c.923T>C | Homozygote | Novel | Leu308Pro |
| 4 | Exon 3 | c.923T>C | Homozygote | Novel | Leu308Pro |
| 5 | Exon 3 | c.923T>C | Heterozygote | Novel | Leu308Pro |
| 6 | Exon 3 | c.923T>C | Heterozygote | Novel | Leu308Pro |
Discussion
CSNB caused by defective signaling from
photoreceptors to bipolar cells is characterized by a reduced or
absent b-wave and a normal a-wave in electroretinography (6). Oguchi disease is an uncommon form of
CSNB, characterized by specific clinical characteristics, termed
the Mizuo-Nakamura phenomenon (7).
Two genes are known to be involved in Oguchi disease: SAG
and GRK1 gene (7). In a
European study, all the reported patients had mutations in
GRK1. However, the majority of Japanese patients with Oguchi
disease have another causative gene, SAG (8).
SAG, also termed arrestin, encodes a major
protein of the outer retinal segment. It binds to activated
rhodopsin in retinal rod outer segments. Mutations in this gene
have previously been associated with Oguchi disease (9). In the present study a novel missense
mutation was identified.
The GRK1 gene encodes rhodopsin kinase,
responsible for the phosphorylated rhodopsin in rod cells (10). Mutations in GRK1 can result
in the Oguchi phenotype. This study demonstrated that a novel amino
acid change (p.Leu308Pro) was common in all siblings in the family
in a homozygous state and thus may have resulted in Oguchi
disease.
To date, in the Turkish population there have been
no reports of GRK1 mutations causing Oguchi disease. In the
present study, a novel nonsense mutation in GRK1 in affected
members of a consanguineous Turkish family was identified. In
conclusion, a novel GRK1 variant (c.923T>C) was shown the
be the cause of Oguchi disease in 4 family members who inherited
the mutation from a common ancestor of their parents.
References
|
1
|
Malaichamy S, Sen P, Sachidanandam R,
Arokiasamy T, Lancelot ME, Audo I, Zeitz C and Soumittra N:
Molecular profiling of complete congenital stationary night
blindness: A pilot study on an Indian cohort. Mol Vis. 20:341–351.
2014.PubMed/NCBI
|
|
2
|
Yamamoto S, Sippel KC, Berson EL and Dryja
TP: Defects in the rhodopsin kinase gene in the Oguchi form of
stationary night blindness. Nat Genet. 15:175–178. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yamamoto H, Simon A, Eriksson U, Harris E,
Berson EL and Dryja TP: Mutations in the gene encoding 11-cis
retinol dehydrogenase cause delayed dark adaptation and fundus
albipunctatus. Nat Genet. 22:188–191. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Waheed NK, Qavi AH, Malik SN, Maria M,
Riaz M, Cremers FP, Azam M and Qamar R: A nonsense mutation in
S-antigen (p.Glu306*) causes Oguchi disease. Mol Vis.
18:1253–1259. 2012.
|
|
5
|
Shimauchi-Matsukawa Y, Aman Y, Tachibanaki
S and Kawamura S: Isolation and characterization of visual pigment
kinase-related genes in carp retina: Polyphyly in GRK1 subtypes,
GRK1A and 1B. Mol Vis. 11:1220–1228. 2005.
|
|
6
|
Van Genderen MM, Bijveld MM, Claassen YB,
Florijn RJ, Pearring JN, Meire FM, McCall MA, Riemslag FC, Gregg
RG, Bergen AA and Kamermans M: Mutations in TRPM1 are a common
cause of complete congenital stationary night blindness. Am J Hum
Genet. 85:730–736. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang L, Li W, Tang W, Zhu X, Ou-Yang P
and Lu G: A Chinese family with Oguchi's disease due to compound
heterozygosity including a novel deletion in the arrestin gene. Mol
Vis. 18:528–536. 2012.PubMed/NCBI
|
|
8
|
Oishi A, Akimoto M, Kawagoe N, Mandai M,
Takahashi M and Yoshimura N: Novel mutations in the GRK1 gene in
Japanese patients With Oguchi disease. Am J Ophthalmol.
144:475–477. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Goldstein O, Jordan JA, Aguirre GD and
Acland GM: A non-stop S-antigen gene mutation is associated with
late onset hereditary retinal degeneration in dogs. Mol Vis.
19:1871–1884. 2013.PubMed/NCBI
|
|
10
|
Azam M, Collin RW, Khan MI, Shah ST,
Qureshi N, Ajmal M, den Hollander AI, Qamar R and Cremers FP: A
novel mutation in GRK1 causes Oguchi disease in a consanguineous
Pakistani family. Mol Vis. 15:1788–1793. 2009.PubMed/NCBI
|