Introduction
Distal hereditary motor neuropathies (dHMN) are a
genetically and clinically heterogeneous group of lower motor
neuron diseases (1). dHMN type 7B
(dHMN7B), which is caused by a mutation in the dynactin 1
(DCTN1) gene, is a late-onset disease characterized by
respiratory difficulties due to bilateral vocal cord palsy,
progressive facial weakness and muscle atrophy in the hands
(2–4). In addition to dHMN7B, mutations in
the DCTN1 gene are known to cause diverse neurodegenerative
diseases, including dHMN7B, Perry syndrome (PS), amyotrophic
lateral sclerosis (ALS) and ALS-frontotemporal dementia (ALS/FTD)
(5–8). PS is an upper motor neuron disease
characterized by Parkinsonism, psychiatric changes, weight loss and
abnormal hypoventilation (8). ALS
is characterized by the combined phenotypes of upper and lower
motor neuron disorders (6).
ALS/FTD presents with progressive changes in personality, behavior
and language, with relative preservation of perception and memory,
due to degeneration of the frontal and temporal lobes of brain
(7).
The DCTN1 gene encodes the largest subunit of
dynactin to provide commands for the synthesis of dynactin 1
(9–12). Dynactin 1 is important for the
retrograde axonal transport of vesicles and organelles along
microtubules (1,13,14).
It has been suggested that dysfunction of dynactin-mediated
transport in the peripheral nerves may lead to motor neuron
disease, and that mutation of the dynein-dynactin microtubule motor
proteins leads to late-onset motor neuron disease in mice (15,16).
The first dHMN7B patient with a DCTN1
mutation was described by Puls et al (5). To the best of our knowledge, no
further cases have been reported. Thus, clinical information about
dHMN7B resulting from a DCTN1 mutation has been limited to a
single family. In the present study, 24 Korean dHMN families were
screened using whole exome sequencing, which identified a
DCTN1 mutation (p.G59S) in two unrelated families. In
addition, the clinical and electrophysiological manifestations of
the dHMN7B patients were described.
Materials and methods
Patients
The DCTN1 gene was sequenced in 62
individuals from 24 families with dHMN. Additionally, 300 Korean
healthy controls with no clinical features or family history of
peripheral neuropathies were recruited from the neurological
department. Written informed consent was obtained from all
individuals, according to the protocol approved by the
Institutional Review Board for Sungkyunkwan University, Samsung
Medical Center (Seoul, Korea).
Clinical assessments
Clinical evaluations were performed by neurologists
and information on deceased family members was obtained from
relatives. Clinical assessments included those for motor
impairments, sensory loss, deep tendon reflexes and vocal cord
palsy. Patient's age at onset was determined by questioning the
patients regarding the first appearance of symptoms. Muscle
strength of flexor and extensor muscles was measured manually using
the standard Medical Research Council scale (17), physical disability was determined
by the functional disability scale (18) and sensory impairment was assessed
based on the level and severity of pain, temperature, vibration and
position.
Electrophysiological study
Motor and sensory conduction velocities were
measured on the median, ulnar, peroneal, tibial and sural nerves.
Motor nerve conduction velocities (MNCVs) of the median and ulnar
nerves were determined by stimulating at the elbow and wrist while
recording compound muscle action potentials (CMAPs) over the
abductor pollicis brevis and adductor digiti quinti, respectively.
Similarly, the MNCVs of the peroneal and tibial nerves were
determined by stimulating at the knee and ankle, while recording
CMAPs over the extensor digitorum brevis and adductor hallucis,
respectively. Sensory nerve conduction velocities were obtained
over a finger-wrist segment from the median and ulnar nerves by
orthodromic scoring, and in addition were recorded for the sural
nerves. Sensory nerve action potential amplitudes were measured
from positive to negative peaks.
DNA preparation and 17p12 duplication
analysis
DNA was purified from peripheral blood samples using
a QIAamp Blood DNA Purification kit (Qiagen GmbH, Hilden, Germany).
All patient samples were prescreened for 17p12 [peripheral
myelin protein 22 (PMP22)] duplication and/or deletion,
which is the most common genetic cause of peripheral neuropathies,
using hexaplex microsatellite PCR analysis, as previously described
(19).
Exome sequencing and identification of
causative mutation
Whole exome sequencing (WES) was performed on two
affected patients from two unrelated families. The exome was
captured using the Human SeqCap EZ Human Exome Library kit (version
3.0; Roche NimbleGen, Inc., Madison, WI, USA), and subsequent
sequencing was performed using a HiSeq 2500 Genome Analyzer
(Illumina, Inc., San Diego, CA, USA). The University of California,
Santa Cruz UCSC assembly hg19 (National Center for Biotechnology
Information build 37.1; genome.ucsc.edu/) was used as the reference sequence
with Burrows-Wheeler Aligner software version 07.11 (bio-bwa.
sourceforge.net/). Using the Sequence
Alignment/Map tools program (samtools.sourceforg.net/), variants were confirmed if
the single nucleotide polymorphism (SNP) qualities were ≥20.
Functionally significant variants, including missense, nonsense,
exonic indel and splicing site variants, were identified in ~70
peripheral neuropathy-associated genes. The sequencing variants
were confirmed by the Sanger sequencing method using an ABI 3130xl
genetic analyzer (Applied Biosystems; Thermo Fisher Scientific,
Waltham, MA, USA) (20). A
mutation was considered as causative when it was identified in
affected individuals and not in the 300 healthy control
samples.
In silico analysis
The conversion pattern for the protein amino acid
sequence was performed in Molecular Evolutionary Genetics Analysis
5 software (version 5.05; www.megas-oftware.net/) (21) and the effect of amino acid
substitution on protein function was predicted using the online
tools of Sorting Intolerant From Tolerant (SIFT; sift.jcvi.org/), Polymorphism Phenotyping version 2
(PolyPhen-2; genetics. bwh.harvard.edu/pph2/) and MUpro (mupro.proteomics.ics.uci.edu/). Genomic
evolutionary rate profiling (GERP) score was determined using the
GERP++ program (mendel.stan-ford.edu/SidowLab/downloads/gerp/index.html).
The domain structure of DCTN1 protein was predicted using the
Simple Modular Architecture Research Tool version 7 (smart.embl.de/).
Results
Clinical manifestations
The clinical presentations of the two families
reported in the present study and the previously reported American
family (5) are summarized in
Table I. The average age of
disease onset in the FC465 and FC654 families was 51.7±8.5 years
(mean ± standard deviation; range, 43–60 years) and 53.8±5.4 years
(range, 47–62 years), respectively, which indicated a later onset
compared with the previously reported American family, in which the
age at disease onset was 34.3±6.2 years. In the two families
described in the present study, no bulbar symptoms, including
dysphagia, dysarthria and facial muscle weakness, were identified
in affected members. The disease severity appeared milder in
patients of the present study compared with the previously reported
family (5). In all affected FC465
family members, stridor and shortness of breath were the first and
predominant symptoms; however, in FC654 family members, hand muscle
weakness occurred prior to vocal cord palsy. Members of the two
families suffered from atrophy and weakness of the first dorsal
interosseous and thenar muscles; however, the hypothenar muscle
bulk and strength was relatively preserved. All seven affected
family members had undergone arytenoidectomy due to respiratory
difficulty caused by bilateral vocal cord palsy. They had bilateral
foot drop and steppage gait; however, no patient became
wheelchair-bound. Patients from the two families exhibited no
sensory loss or upper motor neuron involvement. Decreased or absent
tendon reflexes were observed in the early stages of the disease,
and pathologic reflexes were not identified. FC465 family members
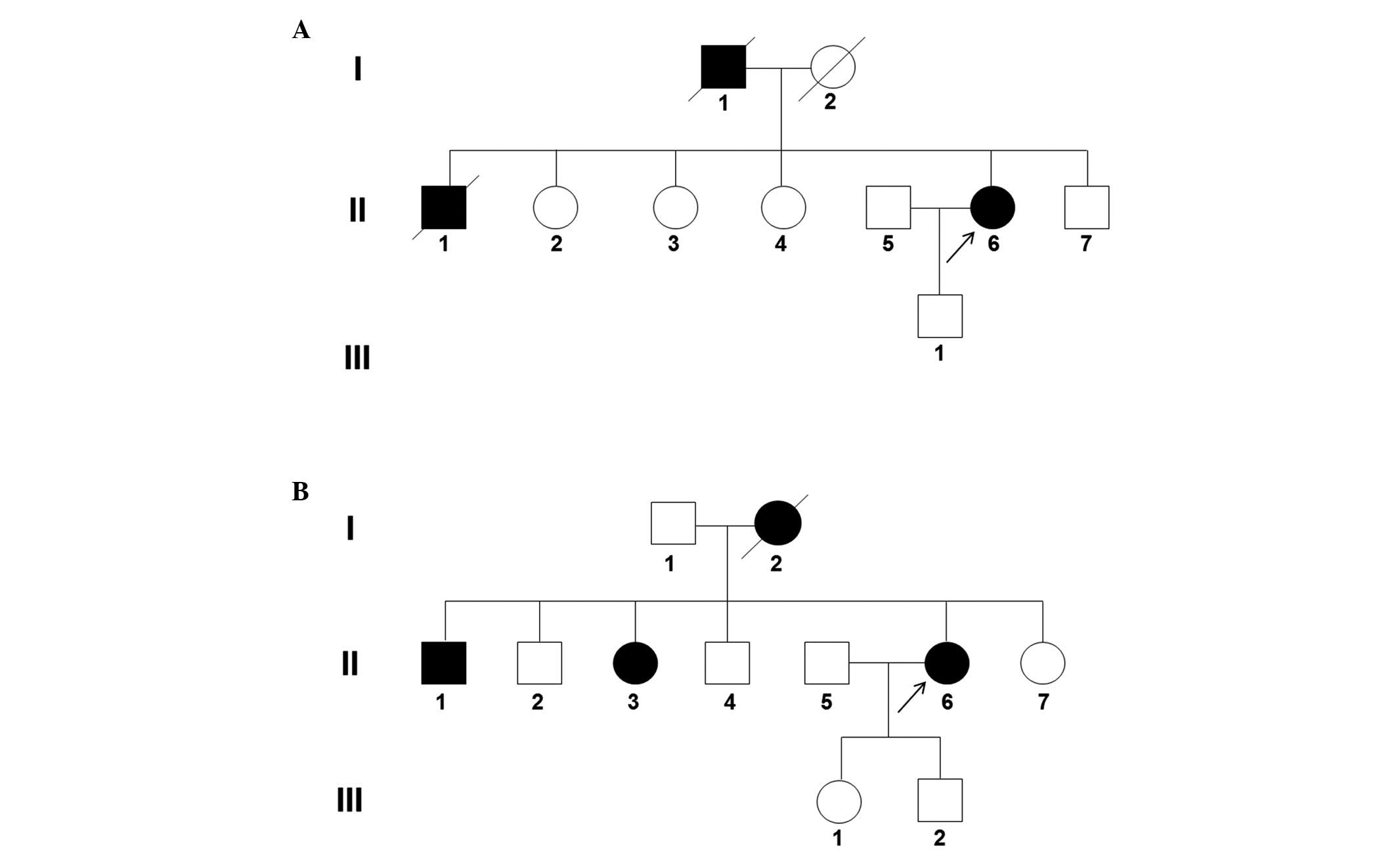
reported that the deceased father (I-1; Fig. 1A) and older brother (II-1; Fig. 1A) had bilateral vocal cord palsy
and had undergone arytenoidectomy, and FC654 family members
reported that the deceased mother (I-2; Fig. 1B) had bilateral vocal cord palsy
and had undergone arytenoidectomy.
 | Table IClinical manifestations of dHMN7B
patients with the dynactin 1 p.G59S mutation. |
Table I
Clinical manifestations of dHMN7B
patients with the dynactin 1 p.G59S mutation.
| Parameter | Korean dHMN7B
families
| American dHMN7B
family |
|---|
| FC465 | FC654 |
|---|
| Number of
patients | 3 | 4 | 9 |
| Inheritance | AD | AD | AD |
| Female ratio, % | 33.3 | 75.0 | 77.8 |
| Age at exam, years
[mean ± SD (range)] | 56.3±7.5 (52–65) | 60.3±4.1 (55–61) | 51.9±8.3 (39–64) |
| Age at onset, years
[mean ± SD (range)] | 51.7±8.5 (43–60) | 53.8±5.4 (47–62) | 34.3±6.2 (23–44) |
| Frequent first
symptom | Stridor | Hand weakness | Stridor |
| FDSa | 2–3 | 2–4 | ND |
| Proximal limb
muscle weakness | None to mild | None to mild | None to mild |
| Distal limb muscle
weakness | Mild to
moderate | Mild to severe | Mild to severe |
| Facial
weakness | No | No | Yes |
| Sensory loss | No | No | No |
| Knee/ankle
jerksb | D, A | A | N, D, A |
| Vocal cord
palsy | Yes | Yes | Yes |
|
Arytenoidectomy | Yes | Yes | Yes |
| Dysphagia | No | No | Yes |
| Dyspnea | Yes | Yes | Yes |
| Foot drop | Bilateral | Bilateral | Bilateral |
|
Wheelchair-bound | No | No | No |
| Tongue
fasciculation | No | No | No |
| Frontotemporal
dementia | No | No | No |
| Parkinsonism | No | No | No |
| Reference | The present
study | The present
study | Puls et al
(2003) (5) |
Pure hereditary motor neuropathy
The nerve conduction study data are summarized in
Table II. The amplitude of the
motor response from the thenar muscles was markedly reduced or
absent in patients 1 [FC465 (II-6)] and 2 [FC654 (II-6)]; however,
the amplitude of the response from the hypothenar muscles were
minimally reduced. Patient 1 had reduced amplitudes of peroneal
innervated foot muscles; however, these responses were reduced
compared with the responses from the thenar muscles. Needle
electromyography in the two patients was consistent with chronic
denervation, and there were scattered fibrillations in distal limb
muscles.
| Table IIElectrophysiology in patients with
the dynactin 1 p.G59S mutation. |
Table II
Electrophysiology in patients with
the dynactin 1 p.G59S mutation.
A, Motor nerve
conduction velocities: Median motor nerve
|
|---|
| Parameter | Patient FC465
(II-6)
| Patient FC654
(II-6)
| Normal value |
|---|
| Right side | Left side | Right side | Left side |
|---|
| TL, ms | A | 3.8 | A | A | <3.9 |
| CMAP, mV | A | 1.2 | A | A | >6.0 |
| MNCV, m/s | A | 35.1 | A | A | >50.5 |
B, Motor nerve
conduction velocities: Ulnar motor nerve
|
|---|
| Parameter | Patient FC465
(II-6)
| Patient FC654
(II-6)
| Normal value |
|---|
| Right side | Left side | Right side | Left side |
|---|
| TL, ms | 2.2 | 2.6 | 2.5 | 3.0 | <3.0 |
| CMAP, mV | 8.9 | 12.5 | 7.5 | 3.8 | >8.0 |
| MNCV, m/s | 54.5 | 55.6 | 52.2 | 64.2 | >51.1 |
C, Motor nerve
conduction velocities: Peroneal nerve
|
|---|
| Parameter | Patient FC465
(II-6)
| Patient FC654
(II-6)
| Normal value |
|---|
| Right side | Left side | Right side | Left side |
|---|
| TL, ms | 3.7 | 3.8 | 4.0 | 3.8 | <5.3 |
| CMAP, mV | 4.7 | 4.4 | 0.6 | 0.1 | >1.6 |
| MNCV, m/s | 43.2 | 43.9 | 48.1 | 43.7 | >41.2 |
D, Motor nerve
conduction velocities: Tibial nerve
|
|---|
| Parameter | Patient FC465
(II-6)
| Patient FC654
(II-6)
| Normal value |
|---|
| Right side | Left side | Right side | Left side |
|---|
| TL, ms | 4.0 | 4.3 | 3.9 | 3.3 | <5.4 |
| CMAP, mV | 8.5 | 10.5 | 11.2 | 10.8 | >6.0 |
| MNCV, m/s | 42.7 | 41.9 | 48.5 | 47.8 | >41.1 |
E, Sensory nerve
conduction velocities: Median sensory nerve
|
|---|
| Parameter | Patient FC465
(II-6)
| Patient FC654
(II-6)
| Normal value |
|---|
| Right side | Left side | Right side | Left side |
|---|
| SNAP,
µV | 11.9 | 29.2 | 55.6 | 43.4 | >8.8 |
| SNCV, m/s | 41.7 | 40.0 | 47.1 | 46.0 | >39.3 |
F, Sensory nerve
conduction velocities: Ulnar sensory nerve
|
|---|
| Parameter | Patient FC465
(II-6)
| Patient FC654
(II-6)
| Normal value |
|---|
| Right side | Left side | Right side | Left side |
|---|
| SNAP,
µV | 8.9 | 22.3 | 35.6 | 12.8 | >7.9 |
| SNCV, m/s | 44.6 | 41.7 | 44.4 | 40.6 | >37.5 |
G, Sensory nerve
conduction velocities: Sural nerve
|
|---|
| Parameter | Patient FC465
(II-6)
| Patient FC654
(II-6)
| Normal value |
|---|
| Right side | Left side | Right side | Left side |
|---|
| SNAP,
µV | 30.7 | 24.7 | 22.4 | 24.0 | >6.0 |
| SNCV, m/s | 34.1 | 35.6 | 44.7 | 47.2 | >32.1 |
Identification of the DCTN1 mutation
Copy number variation on 17p12 (PMP22) was
not observed; therefore WES was performed on the affected proband
in each family. The mean total WES yield was ~12.7 Gbp/sample with
a 94.37% coverage rate (≥10X read depth) for the target regions.
The average number of observed coding variants was ~24,765 SNPs
including insertion/deletion variants.
By filtering of the functionally significant
variants in >70 peripheral neuropathy-associated genes, the
missense mutation in the DCTN1 gene was identified in two
dHMN families: c.175G>A (p.G59S) in FC465 and FC654 families.
This mutation was confirmed by the Sanger's DNA sequencing method
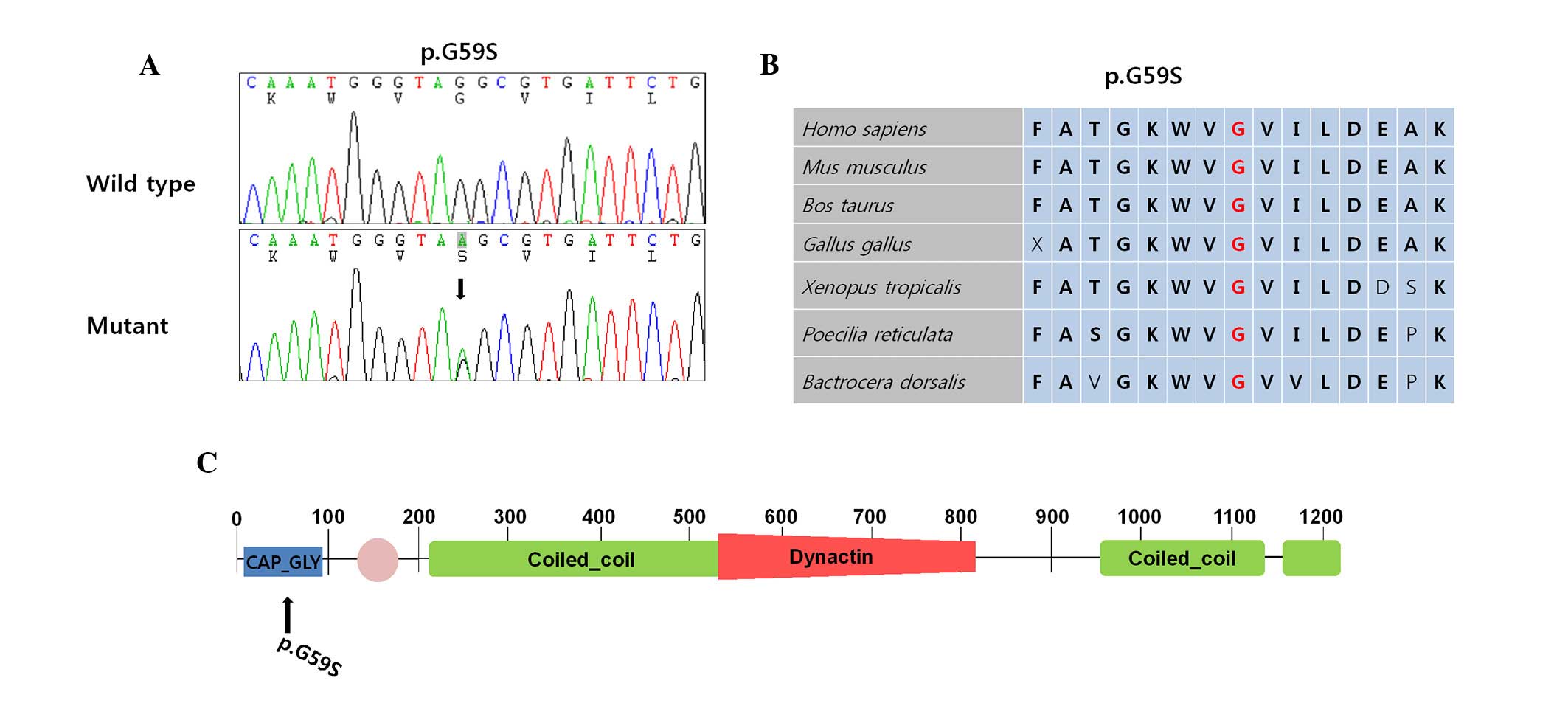
(Fig. 2A). The p.G59S was located
in the cytoskeleton-associated protein glycine-rich domain. This
mutation was not observed in any of the 300 healthy controls, and
it was not reported in the dbSNP144 database (www.ncbi.nlm.nih.gov), the 1000 Genomes project
database (www.1000genomes.org/) or the Exome
Variant Server (evs.gs.washington.edu/EVS/). Conservation analysis
revealed that the vicinity of mutation site was conserved among the
species assessed (Fig. 2B).
The p.G59S mutation has been previously reported to
be the underlying cause of dHMN7B (5). Various causative mutations for other
neurodegenerative diseases were located in the coiled-coil domain
(Fig. 2C) (7,8). The
GERP score of 5.14 was considerably high in the two mutated
nucleotide sequences, and in silico analyses using SIFT,
PolyPhen-2 and MUpro predicted the affected function of the mutant
protein (Table III). Although
other non-synonymous variants were identified in the peripheral
neuropathy-associated genes, with the exception of the DCTN1
mutations, no other variants were considered as the underlying
cause of dHMN, as they were identified in healthy controls.
| Table IIIIn silico analysis of
dynactin 1 mutations. |
Table III
In silico analysis of
dynactin 1 mutations.
| In silico
analysis | c.175G>A
(p.G59S) | Prediction |
|---|
| SIFT | 0.000a | Affection of
protein function |
| PolyPhen-2 | 1.000a | Damaging |
| MUpro | −0.880a | Decreased of
structure stability |
Discussion
In the present study, a p.G59S mutation was
identified as the underlying cause of dHMN7B in two families, and
their detailed clinical features were characterized. This is the
second description of dHMN7B resulting from a DCTN1
mutation, following the initial report in 2003 (5). Codon 59 appears to be the mutational
hot spot in the DCTN1 gene, as all described dHMN7B patients
to date have harbored identical p.G59S mutations.
The families with the DCTN1 mutation reported
in the present study had similar clinical features to the
previously described family. All patients underwent an
arytenoidectomy to provide an adequate airway and hand muscle
weakness affected the thenar more than the hypothenar muscles. All
patients had steppage gait, but none became wheelchair-bound.
However, clinical heterogeneities were observed between the present
and previously reported patients. The age of onset in the FC465 and
FC654 families was later compared with the previously reported
American family. No affected family members of the present study
exhibited any bulbar symptoms, including facial weakness, dysphagia
and dysarthria. The severity of the disease manifestations was
milder compared with that in the previously reported family. In
addition, interfamilial differences were observed. In affected
FC465 members, respiratory difficulty during exercise was the
initial and predominant symptom; however, in affected FC654
members, hand muscle weakness was the first dominant symptom. Thus,
diverse clinical features of an identical DCTN1 mutation
were observed; this may be useful for the future diagnosis of
dHMN7B patients with a DCTN1 mutation.
The phenotype of the disease in the patients of the
present study differs from other diseases associated with
DCTN1 mutations, including ALS, ALS/FTD and PS, in important
aspects. The most significant difference is the absence of lower
motor neuron signs in the other diseases. All patients of the
present study exhibited decreased or absent deep tendon reflexes;
however, patients with ALS, ALS/FTD or PS exhibited normal or
increased deep tendon reflexes. Furthermore, the pathognomonic
symptoms of disease in the patients of the present study did not
tend to overlap with ALS, ALS/FTD and PS (Table I).
The frequency of dHMN7B resulting from a
DCTN1 mutation appears to be extremely low. To the best of
our knowledge, no cases have been reported since the first report
of dHMN7B family with p.G59S DCTN1 mutation (5). However, the present study identified
the p.G59S mutation of the DCTN1 gene in two Korean dHMN
families from 24 unrelated families (8.3%), which is not extremely
rare. It may be that this mutation is relatively common in Asia and
rare in the west. In addition, DCTN1 mutations have been
demonstrated to result in upper motor neuron diseases, including
PS, ALS and ALS/FTD. To date, mutations of the DCTN1 gene
have not generally been considered to cause lower motor neuron
diseases, due to the lack of evidence supporting the initial report
in dHMN7B. In conclusion, the results of the present study
suggested that the DCTN1 mutation may not be extremely rare,
particularly in the Korean population, and therefore screening for
the DCTN1 gene must be considered for all dHMN patients.
Acknowledgments
The authors would like to thank the patients and
their families for their participation. The present study was
supported by the Korean Health Technology R&D Project, Ministry
of Health & Welfare (grant nos. HI12C0135 and HI14C3484) and by
the National Research Foundation of Korea grants funded by the
Ministry of Science, ICT and Future Planning (grant no.
NRF-2014R1A2A2A01004240).
References
|
1
|
Rossor AM, Kalmar B, Greensmith L and
Reilly MM: The distal hereditary motor neuropathies. J Neurol
Neurosurg Psychiatry. 83:6–14. 2012. View Article : Google Scholar
|
|
2
|
Puls I, Oh SJ, Sumner CJ, Wallace KE,
Floeter MK, Mann EA, Kennedy WR, Wendelschafer-Crabb G, Vortmeyer
A, Powers R, et al: Distal spinal and bulbar muscular atrophy
caused by dynactin mutation. Ann Neurol. 57:687–694. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
2nd Workshop of the European CMT
Consortium: 53rd ENMC International Workshop on Classification and
Diagnostic Guidelines for Charcot-Marie-Tooth Type 2 (CMT2-HMSN II)
and Distal Hereditary Motor Neuropathy (distal HMN-Spinal CMT)
26–28 September 1997, Naarden, The Netherlands. Neuromuscul Disord.
8:426–431. 1998. View Article : Google Scholar
|
|
4
|
Harding AE and Thomas PK: Genetic aspects
of hereditary motor and sensory neuropathy (types I and II). J Med
Genet. 17:329–336. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Puls I, Jonnakuty C, LaMonte BH, Holzbaur
EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, et al:
Mutant dynactin in motor neuron disease. Nat Genet. 33:455–456.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Münch C, Sedlmeier R, Meyer T, Homberg V,
Sperfeld AD, Kurt A, Prudlo J, Peraus G, Hanemann CO, Stumm G and
Ludolph AC: Point mutations of the p150 subunit of dynactin (DCTN1)
gene in ALS. Neurology. 63:724–726. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Münch C, Rosenbohm A, Sperfeld AD, Uttner
I, Reske S, Krause BJ, Sedlmeier R, Meyer T, Hanemann CO, Stumm G
and Ludolph AC: Heterozygous R1101K mutation of the DCTN1 gene in a
family with ALS and FTD. Ann Neurol. 58:777–780. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Farrer MJ, Hulihan MM, Kachergus JM,
Dächsel JC, Stoessl AJ, Grantier LL, Calne S, Calne DB, Lechevalier
B, Chapon F, et al: DCTN1 mutations in Perry syndrome. Nat Genet.
41:163–165. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Irobi J, Dierick I, Jordanova A, Claeys
KG, De Jonghe P and Timmerman V: Unraveling the genetics of distal
hereditary motor neuronopathies. Neuromolecular Med. 8:131–146.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Holzbaur EL and Vallee RB: DYNEINS:
Molecular structure and cellular function. Annu Rev Cell Biol.
10:339–372. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Irobi J, De Jonghe P and Timmerman V:
Molecular genetics of distal hereditary motor neuropathies. Hum Mol
Genet. 13:R195–R202. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Moore JK, Sept D and Cooper JA:
Neurodegeneration mutations in dynactin impair dynein-dependent
nuclear migration. Proc Natl Acad Sci USA. 106:5147–5152. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Levy JR, Sumner CJ, Caviston JP, Tokito
MK, Ranganathan S, Ligon LA, Wallace KE, LaMonte BH, Harmison GG,
Puls I, et al: A motor neuron disease-associated mutation in
p150Glued perturbs dynactin function and induces protein
aggregation. J Cell Biol. 172:733–745. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vaughan KT and Vallee RB: Cytoplasmic
dynein binds dynactin through a direct interaction between the
intermediate chains and p150Glued. J Cell Biol. 131:1507–1516.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
LaMonte BH, Wallace KE, Holloway BA,
Shelly SS, Ascaño J, Tokito M, Van Winkle T, Howland DS and
Holzbaur EL: Disruption of dynein/dynactin inhibits axonal
transport in motor neurons causing late-onset progressive
degeneration. Neuron. 34:715–727. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hafezparast M, Klocke R, Ruhrberg C,
Marquardt A, Ahmad-Annuar A, Bowen S, Lalli G, Witherden AS,
Hummerich H, Nicholson S, et al: Mutations in dynein link motor
neuron degeneration to defects in retrograde transport. Science.
300:808–812. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Paternostro-Sluga T, Grim-Stieger M, Posch
M, Schuhfried O, Vacariu G, Mittermaier C, Bittner C and
Fialka-Moser V: Reliability and validity of the Medical Research
Council (MRC) scale and a modified scale for testing muscle
strength in patients with radial palsy. J Rehabil Med. 40:665–671.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Birouk N, Gouider R, Le Guern E, Gugenheim
M, Tardieu S, Maisonobe T, Le Forestier N, Agid Y, Brice A and
Bouche P: Charcot-Marie-Tooth disease type 1A with 17p11.2
duplication. Clinical and electrophysiological phenotype study and
factors influencing disease severity in 119 cases. Brain.
120:813–823. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Choi BO, Kim J, Lee KL, Yu JS, Hwang JH
and Chung KW: Rapid diagnosis of CMT1A duplications and HNPP
deletions by multiplex microsatellite PCR. Mol Cells. 23:39–48.
2007.PubMed/NCBI
|
|
20
|
Sanger F, Nicklen S and Coulson AR: DNA
sequencing with chain-terminating inhibitors. Proc Natl Acad Sci
USA. 74:5463–5467. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tamura K, Peterson D, Peterson N, Stecher
G, Nei M and Kumar S: MEGA5: Molecular evolutionary genetics
analysis using maximum likelihood, evolutionary distance, and
maximum parsimony methods. Mol Biol Evol. 28:2731–2739. 2011.
View Article : Google Scholar : PubMed/NCBI
|