Introduction
Sphingosine-1-phosphate receptor 1 (S1PR1) belongs
to the G protein-coupled receptor family for sphingosine
1-phosphate (S1P), a biologically active lipid. S1P-S1PR1 signaling
is vital in a variety of developmental processes, including cell
growth, survival and migration (1). However, S1PR1 signaling also promotes
tumor progression and tumor angiogenesis (2,3). In
myeloid cells derived from tumors, including activated B cell-like
diffuse large B-cell lymphoma, the expression of S1PR1 is elevated,
compared with normal splenic myeloid cells, and this upregulation
is correlated with the persistent activation of signal transducer
and activator of transcription 3 (STAT3) (2,3).
S1PR1-STAT3 signaling is also crucial for myeloid cell colonization
at future metastatic sites (4).
Previous data has suggested a correlation between reduced S1PR1 and
reduced survival rates of patients with glioblastoma (5), and the high level of S1PR1 in
T-lymphoblastic lymphoma (T-LBL) is a key factor in the inhibition
of progression of T-LBL to acute T-lymphoblastic leukemia (6). Studies based on certain S1PR1
inhibitors have also suggested that the degradation of S1PR1 can
arrest cell growth in various cancer cell lines (6–8).
Cell growth and apoptosis involve several signaling
pathways. The direct executors of apoptosis are intracellular
cysteine proteases, termed caspases (9). Caspases are originally translated as
inactive zymogens, and subsequently activated by 'mitochondria' or
the 'death receptor' pathway. Of note, the release of cytochrome
c from mitochondria, which triggers caspase activation,
appears to be predominantly mediated by the direct or indirect
actions of reactive oxygen species (ROS) (10). The response of tumor cells to cell
death stimuli is a consequence of cellular redox status, and
stimuli, which induce generation of intracellular ROS facilitate
the execution of cell death (11).
ROS, including hydroxyl, superoxide, nitroxyl radicals, hydrogen
peroxide, organic hydroperoxides and hypochlorous acid, can destroy
DNA and proteins, and activate cell death (11,12).
ROS generation has been shown to be associated with the activation
of c-Jun N-terminal kinase (JNK)/stress-activated protein kinase by
oxidative inactivation of endogenous JNK inhibitors, including JNK
phosphatase receptor-mediated cell death (13–15).
JNK and other members of the mitogen-activated protein kinase
(MAPK) family, including extracellular signal-regulated kinase
(ERK)1/2 and p38 MAPK, are involved in cell proliferation and
apoptosis through the activation of specific transcription
factors.
With the lowest survival rate among the four major
types of leukemia, acute myeloid leukemia (AML) is a significant
threat to human health (16).
Clinically, AML is characterized as the abnormal differentiation
and proliferation of immature blast cells in the bone marrow, and
is known to be the most common form of acute leukemia among adults
(16). However, the pathogenesis
of the disease remains to be elucidated.
Although several studies have suggested that S1PR1
is a notable factor for solid tumors, the role of S1PR1 in AML cell
growth remains to be fully elucidated (17). In the present study, the effects of
S1PR1 in modulating the apoptosis and proliferation of AML cells
were investigated. It was found that S1PR1 signaling mediated
anti-apoptotic/pro-proliferative processes in the AML cells through
the inhibition of mitochondrion-associated apoptosis and ROS
generation, and via the regulation of multiple MAPK signaling
cascades.
Materials and methods
Cell culture and reagents
The human promyelocytic leukemia cell lines, HL60,
U937 and K562, were purchased from American Type Culture Collection
(Manassas, CA, USA) and propagated in RPMI 1640 (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) in
a 37°C incubator with 5% CO2. The 2-7-dichlorofluorescin
diacetate (DCFH-DA) and N-acetyl-L-cysteine (NAC) were purchased
from Sigma-Aldrich (MO, St. Louis, MA, USA) and dissolved in saline
solution at pH 7.0.
Overexpression and knockdown of
S1PR1
cDNA was synthesized from RNA using a cDNA synthesis
kit (Thermo Fisher Scientific, Inc.). Prior to reverse
transcription, RNA was treated with DNase (Invitrogen; Thermo
Fisher Scientific, Inc.). The coding sequence of S1PR1 was obtained
by polymerase chain reaction (PCR), using cDNA generated from the
RNA of HL60 as a template, with the following primers: S1PR1,
forward 5′-ATGGATGACCTAATCGCCAGACAC-3′ and reverse
5′TTAAGAAGAAGAGGTGATATT-3′. The coding sequence was annealed and
digested using AgeI and EcoRI, and then was ligated
into pCDNA3 (Invitrogen; Thermo Fisher Scientific, Inc.). The
recombinant plasmid was verified by direct sequencing and used to
induce the overexpression of S1PR1 in the HL60 cell lines. The
siRNA sequence target for S1PR1 was 5′-ATGATCGATCATCTATAGCAA-3′
(18), and a non-silencing small
interfering (si) RNA was used as a control (Life Technologies,
Grand Island, NY, USA). For transfection and the analysis of
expression levels, a total of 1×106 HL60 cells were
suspended in 1 ml OPTI-MEM and subsequently transfected with 100 pM
siRNA or 4 µg pCDNA3-S1PR1 using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.). After 48 h, the
expression of S1PR1 was analyzed by quantitative (q)PCR using a
SYBR Green PCR kit (Toyobo, Tokyo, Japan) with the 7300 Real-Time
PCR system (Applied Biosystems, Darmstadt, Germany). Expression
levels were normalized to GAPDH and measured in triplicate. The
following primers were used: S1PR1, forward
5′-CCTCGGTGGTGTTCATTC-3′ and reverse 5′-GCAGGTTAGCTGTGTAGG-3′;
GAPDH, forward 5′-CACCCACTCCTCCACCTTTG-3′ and reverse
5′-CCACCACCCTGTTGCTGTAG-3′. The PCR cycling conditions were as
follows: 95°C for 10 min, followed by 40 cycles at 95°C for 15 sec
and 60°C for 45 sec, and a final extension step of 95°C for 15 sec,
60°C for 1 min, 95°C for 15 sec and 60°C for 15 sec. The gene
expression was calculated using the 2−ΔΔCq method
(19).
Western blot analysis
Whole cell protein extracts were isolated using RIPA
buffer containing 50 mM Tris-HCl, (pH 8.0), 150 mM NaCl, 1% Nonidet
P-40, 0.1% SDS, 2 mM phenylmethylsulfonyl fluoride, phosphatase and
protease inhibitor cocktail (CalbioChem, San Diego, CA, USA) at 4°C
for 20 min, followed by centrifugation at 12,000 g for 1 min at
25°C. The supernatants were then subjected to SDS-PAGE (Bio-Rad
Laboratories, Inc., Richmond, CA, USA). The protein samples were
consequently transferred onto PVDF membrane (0.22 µm; EMD
Millipore, Billerica, MA, USA) using a wet transfer device (Bio-Rad
Laboratories, Inc.), followed by blotting with primary antibodies
specific for B cell lymphoma-2 (Bcl-2)-associated X protein (Bax;
cat. no, Sc-493), Bcl2 (cat. no. Sc-492; Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA), Caspase 3 (cat. no. Ab32351),
phosphorylated (p)-c-Jun (cat. no Ab32385), Jun (cat. no. Ab32137),
MKK1 (cat. no. Ab139343), p-MKK1 (cat. no. Ab3338) from Abcam, and
p38 (8690), p-p38 (cat. no. 4631), ERK (cat. no. 9102), p-ERK (cat.
no. 5726), JNK (cat. no. 9258), p-JNK (cat. no. 5291), GAPDH (cat.
no. 5174) from Cell Signaling Technology, Inc. (Beverly, MA, USA)
for 2 h at 25°C, prior to incubation with secondary antibodies for
1 h at 37°C, and were subsequently washed three times with
Tris-buffered saline with 20% Tween-20 (Amresco, LLC, Solon, OH,
USA). The secondary antibodies used were horseradish
peroxidase-conjugated goat anti-rabbit IgG (cat. no. A0216;
Beyotime Institute of Biotechnology, Shainghai, China) or
horseradish peroxidase-conjugated goat anti-mouse IgG (cat. no.
A0208; Beyotime Institute of Biotechnology). The bands on the
membrane were developed using an ECL kit (EMD Millipore) and
visualized by exposure to a UVP bio-imaging system (UVP, Inc.,
Upland, CA, USA).
Cell apoptosis and viability assays
Cell apoptosis was detected using a Fluorescein
isothiocyanate (FITC) Annexin V Apoptosis Detection kit with
propidium iodide (PI; BioLegend, Inc., San Diego, CA, USA).
Briefly, the cell samples were washed twice with ice-cold Annexin V
binding buffer, and then stained with Annexin V-FITC and PI for 15
min at room temperature, followed by the addition of another 400
µl Annexin V binding buffer and analysis using flow
cytometry (Accuri C6; BD Biosciences, San Diego, CA, USA). Early
stage apoptotic cells were stained with FITC Annexin V, but not PI,
whereas late stage apoptotic cells and necrotic cells stained
positively for FITC Annexin V and PI. Cell viability was assessed
using a Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto,
Japan), according to the manufacture's protocol. Briefly,
2.5×103 target cells were seeded in triplicate into a 96
well plate, following which 10 µl of
2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium,
monosodium salt (WST-8) solution was added to each well and
incubated for 4 h in the incubator. The viable cell-mediated
formation of WST-8 formazan was measured at an absorbance of 450 nm
using a microplate reader.
Analysis of intracellular ROS levels
To assess the involvement of S1PR1 in the reduction
of ROS, the HL60 cells were transfected with S1PR1 constructs, with
or without pre-treatment with 100 µM NAC for 2 h. After 48
h, the cell samples were incubated with 5 M of the redox-sensitive
dye, DCF-DA, for 30 min at 37°C. The ROS-mediated oxidation of the
fluorescent compound, DCF, was measured by excitation at 488 nm and
an emission wavelength of 525 nm using a flow cytometer (BD
Biosciences).
Statistical analysis
Data are presented as the mean ± standard deviation.
Statistical analysis was performed by two-way analysis of variance
followed by Tukey's test. Statistical analyses were performed using
GraphPad Prism software, version 5 (GraphPad Software, Inc., La
Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Modulation of the expression of S1PR1 by
transient transfection with plasmids
To elucidate the effects of S1PR1 in myeloid
leukemia cell proliferation and apoptosis, the present study first
constructed a mammalian cell expression plasmid for the
overexpression of S1PR1 and an siRNA plasmid for knockdown of the
expression of S1PR1. As shown in the qPCR assay, the mRNA level of
S1PR1 was increased markedly in the HL60 cells transfected with the
S1PR1 expression plasmid, whereas transfection with the siRNA
plasmid led to a significant reduction in the endogenous expression
of S1PR1 (Fig. 1A;
P<0.001).
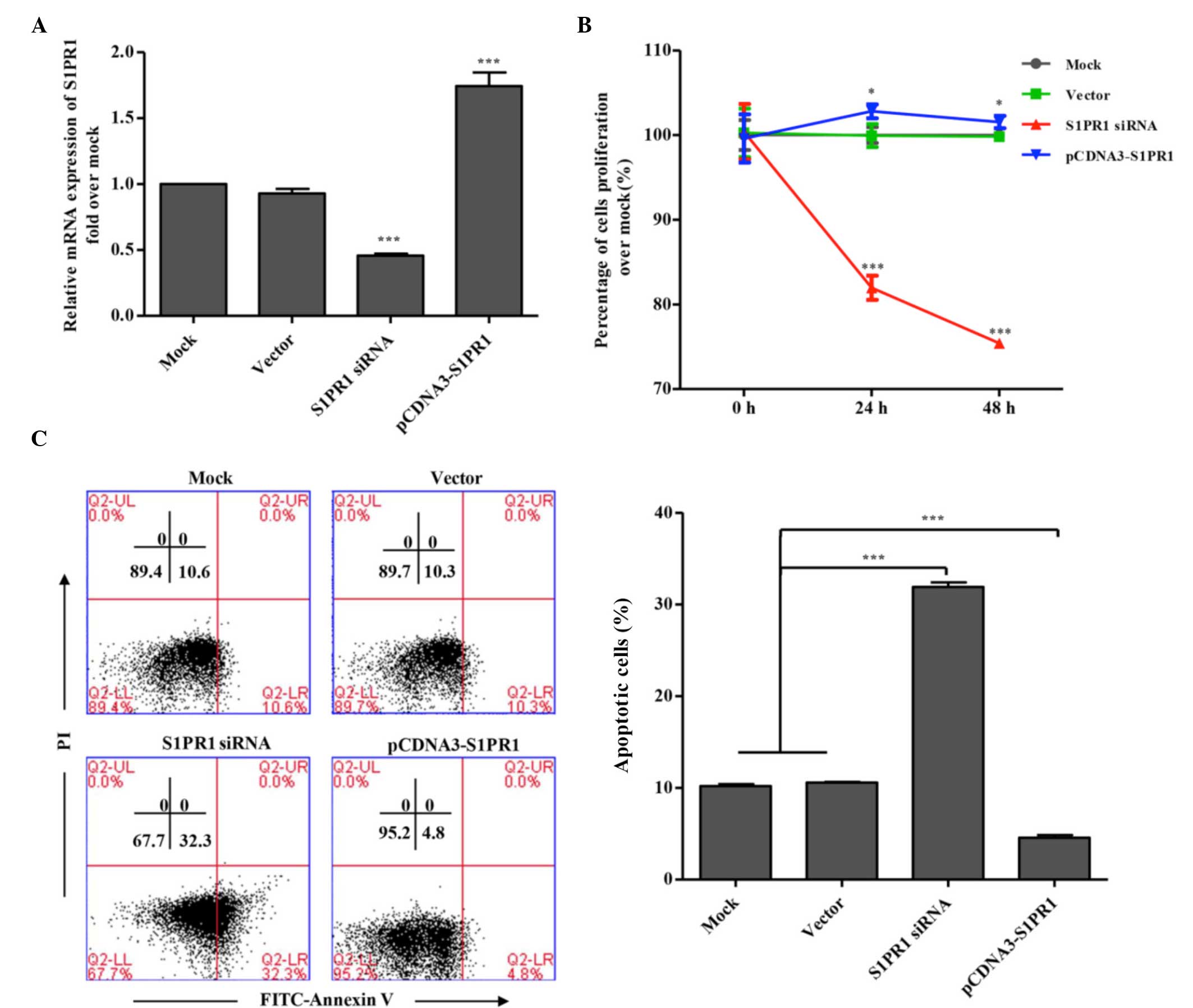 | Figure 1Effects of the expression of S1PR1 on
the proliferation and apoptosis of AML cells. (A) Modulation of the
expression of S1PR1 by transient transfection with plasmids. HL60
cells were transfected with the indicated plasmids. At 48 h
post-transfection, total RNA was isolated from the cell samples and
the mRNA levels of S1PR1 were analyzed using quantitative
polymerase chain reaction analysis. Data are presented as the mean
± standard deviation (n=3). (B) Effects of the expression of S1PR1
on the proliferation of human AML cells. U937 cells were
transfected with the indicated plasmids. Cell proliferation was
measured at the indicated times post-transfection using a CCK-8
assay. Data are presented as the mean ± standard deviation (n=3).
(C) Effects of the expression of S1PR1 on apoptosis of human AML
cells. U937 cells were transfected with S1PR1 siRNA or
overexpression plasmids, or control plasmids. After 48 h, cell
apoptosis was analyzed with Annexin-PI staining using flow
cytometry. A representative assay is shown, with data presented as
the mean ± standard deviation (n=3). *P<0.05 and
***P<0.001, compared with mock and vector. S1PR1,
sphingosine-1-phosphate receptor 1; AML, acute myeloid leukemia;
siRNA, small interfering RNA; FITC, fluorescein isothiocyanate; PI,
propidium iodide. |
S1PR1 inhibits the apoptosis of human
myeloid leukemia cells and promotes their proliferation
The present study examined the effects of the
expression of S1PR1 on the proliferation of human myeloid leukemia
cells using a CCK-8 kit. The U937 cells were transfected with the
siRNA plasmid, the S1RP1 expression plasmid, or the corresponding
control plasmids. Cell proliferation was measured at 0, 24 and 48 h
post-transfection using a CCK-8 assay. As shown in Fig. 1B, knockdown of the expression of
S1PR1 by transfection with the siRNA plasmid significantly
inhibited U937 cell proliferation, and the overexpression of S1PR1
appeared to result in a marginal enhancement of cell proliferation.
To investigate whether S1PR1 contributes to the inhibition of cell
apoptosis, the effect of S1PR1 on cell apoptosis was examined by
Annexin-PI staining and flow cytometry. As shown in Fig. 1C, knockdown of the expression of
S1PR1 by transfection with the siRNA plasmid markedly induced the
apoptosis of leukemia cells, whereas cell apoptosis appeared to be
inhibited by the overexpression of S1PR1, suggesting that S1PR1
signaling likely led to the suppression of apoptosis and the
promotion of proliferation in the human leukemia cells.
S1PR1 confers resistance to
mitochondrion-associated apoptosis in myeloid leukemia cells
The apoptotic process in cells is strictly
controlled by the expression of anti- and pro-apoptotic proteins.
For example, the Bcl-2 family contributes to the regulation of cell
apoptosis by functioning as promoters, including Bax, or
inhibitors, including Bcl-2 of mitochondrion-associated apoptosis.
To further elucidate the molecular mechanism by which S1PR1
suppresses the apoptosis of human myeloid leukemia cells, the
present study assessed the protein expression levels of
pro-apoptotic Bax and anti-apoptotic Bcl-2, in the context of the
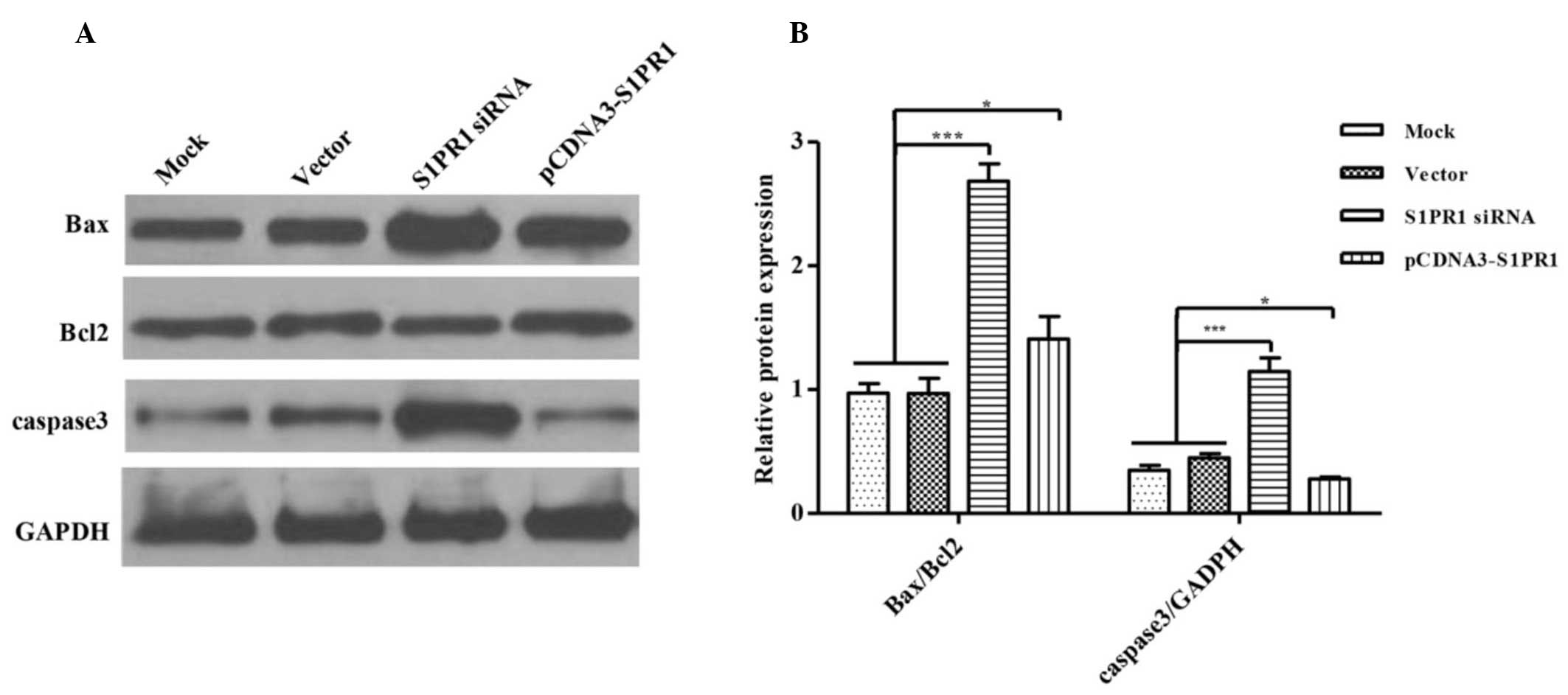
overexpression or downregulation of S1PR1. As shown in Fig. 2, the overexpression of S1PR1
resulted in a significant increase in the expression of Bax, thus
reducing the ratio of Bax/Bcl-2. This suggested that S1PR1 affected
cell apoptosis by regulating the expression of
mitochondrion-associated anti- and pro-apoptotic proteins. In
addition, the effects of the expression of S1PR1 on the activation
of caspase-3 were examined. There was an increase in the level of
cleaved caspase-3 (17 kD) in the cells with downregulated levels of
S1PR1, whereas the overexpression of S1PR1 led to the inhibition of
caspase-3 cleavage (Fig. 2). Taken
together, these data demonstrated that S1PR1 interfered with
myeloid leukemia cell apoptosis via the suppression of
mitochondrion-associated apoptotic processes by regulating the
expression of Bax and activation of caspase-3.
S1PR1 suppresses the generation of
ROS
Several previous studies have shown that ROS is
important in stress-induced apoptosis. During cellular stress, ROS
can act directly on the apoptotic machinery, by accelerating
depolarization and causing dysfunction of mitochondria (20), which in turn, functions as a major
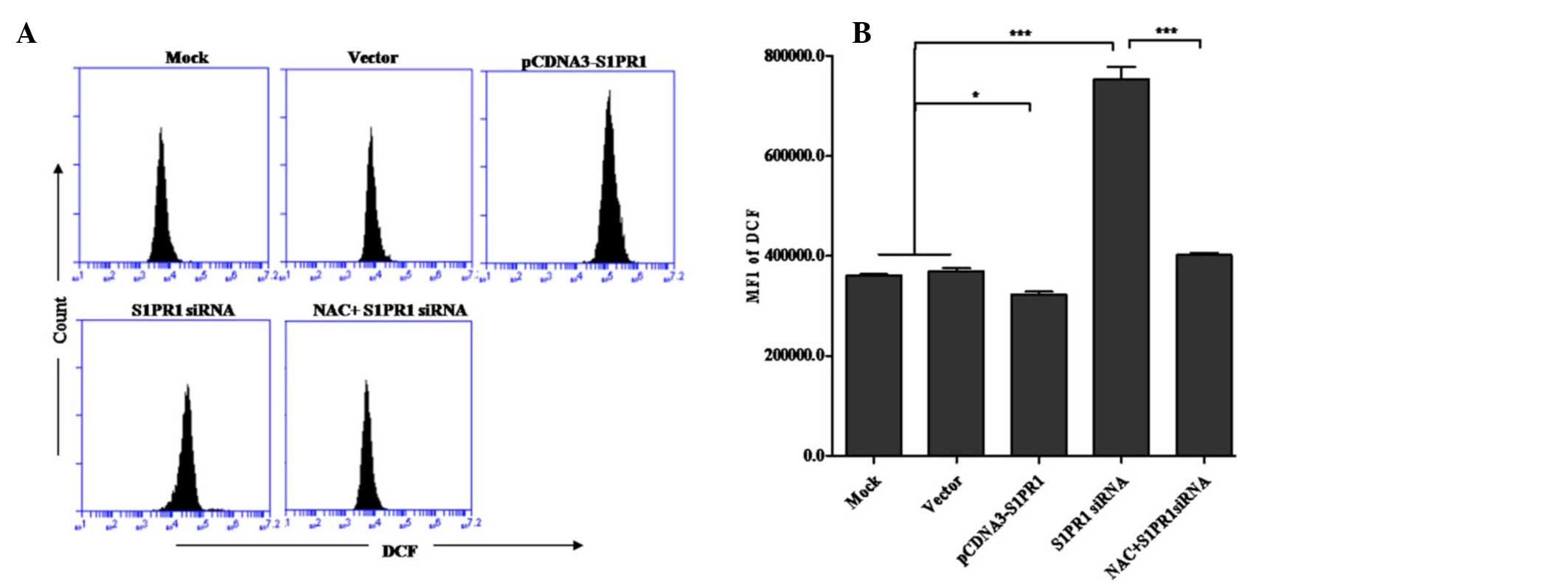
source of ROS in the course of apoptosis (21). To investigate whether ROS is
involved in the regulation of S1PR1-mediated myeloid leukemia cell
apoptosis, the present study compared the levels of ROS in cells
with S1PR1 overexpression or knockdown with the control cells. As
shown in the results of the flow cytometric analysis (Fig. 3), the downregulation of S1PR1 led
to a marked increase in the intracellular levels of ROS. By
contrast, the generation of ROS in cells overexpressing S1PR1 was
significantly suppressed. To further confirm the involvement of ROS
in the S1PR1-mediated regulation of cell apoptosis, NAC, a ROS
scavenging agent was used in flow cytometric analysis of
S1PR1-mediated regulation of cell apoptosis. As expected, cell
apoptosis induced by the downregulation of S1PR1 was partially
reversed in cells treated with NAC, further indicating the direct
implication of ROS in the S1PR1-mediated resistance of cell
apoptosis. These results suggested that S1PR1 confers resistance to
cell apoptosis likely by suppressing the generation or accelerating
the elimination of ROS.
S1PR1 inhibits JNK-c-Jun signaling
Apart from the direct action of ROS on mitochondrial
apoptotic machinery, ROS can also promote apoptosis by triggering
the pro-apoptotic signaling pathway mediated by JNK and p38 MAPK.
As ROS is involved in the regulation of cell apoptosis by S1PR1, it
was hypothesized that the regulation may also involve ROS-induced
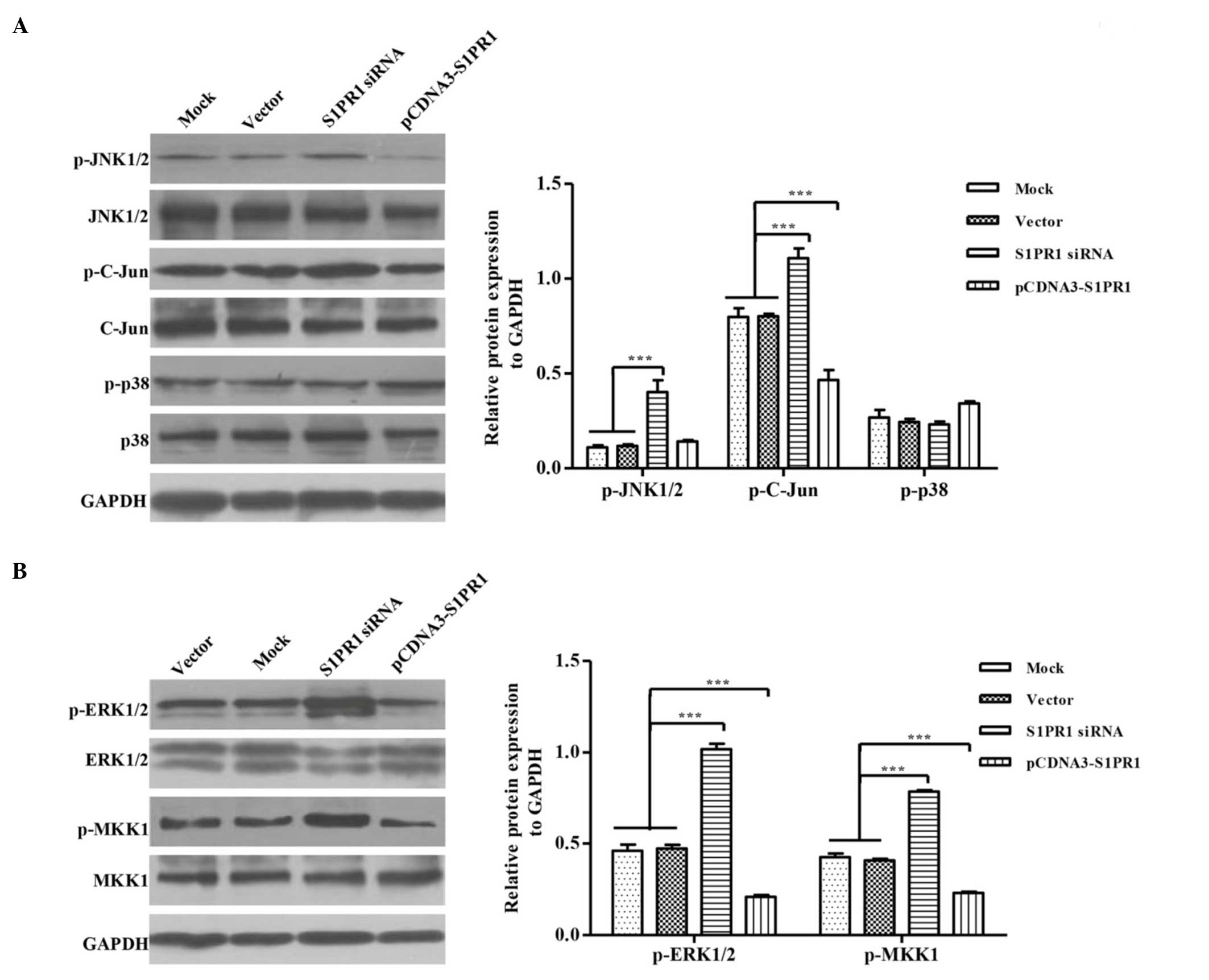
JNK or/and P38 MAPK signaling. To confirm this hypothesis, the
activation of JNK and P38 were examined by western blot analysis
using antibodies against corresponding phosphorylated proteins. The
phosphorylation of p38 was not affected by either the
overexpression or knockdown of S1PR1. However, JNK activation was
not changed in S1PR1-overexpressing cells, and the knockdown of
S1PR1 appeared to induce a significant increase in JNK
phosphorylation, compared with the controls (Fig. 4A), suggesting that the
downregulated expression of S1PR1 contributed to the activation of
JNK. Similar results were obtained from the investigation of the
level of c-Jun phosphorylation downstream of the JNK signaling.
These findings indicated that S1PR1 specifically interfered with
JNK signaling and likely inhibited cell apoptosis.
S1PR1 positively modulates MKK1-ERK
signaling, likely resulting in the promotion of cell
proliferation
In parallel with JNK and P38, ERK is a member of the
MAPK super-family. However, JNK and p38 MAPK are preferentially
activated by proinflammatory cytokines and oxidative stress,
resulting in cell apoptosis, whereas ERK1/2 are activated by
mitogens and growth factors, leading to cell growth and survival.
Thus, the present study evaluated the activation of ERK. The
overexpression of S1PR1 increased the phosphorylation of ERK,
whereas kinase activation appeared to be marginally attenuated in
the S1PR1-knockdown cells (Fig.
4B). These results indicated that S1PR1 had a positive effect
in the activation of ERK. In addition, the effects of the
expression of S1PR1 on the phosphorylation of MKK1, the upstream
kinase of ERK, was also examined and a similar result was obtained,
suggesting that the enhancement of ERK activation was due to the
positive regulation of MKK1 signaling by S1PR1. Considering the
roles of ERK signaling in cell growth and survival, it was
hypothesized that the promotion of cell proliferation observed
results, at least in part, from the positive modulation of MKK1-ERK
signaling by S1PR1. Taken together, S1PR1 likely suppresses JNK
signaling, leading to the promotion of myeloid leukemia cell
apoptosis, and enhances cell proliferation by activating ERK
signaling.
In conclusion, S1PR1 not only suppressed JNK
signaling and promoted myeloid leukemia cell
(mitochondrion-associated) apoptosis, but also facilitated cell
proliferation, likely by suppressing the generation or accelerating
the elimination of ROS, and by activating ERK signaling.
Discussion
AML is a heterogeneous clonal malignancy. The
disease is characterized as multiple genetic alterations in
hematopoietic stem cells, including the translocation of a t(12;18)
(p13;q12) involving Ets Variant 6, which results in the
overexpression of SET binding protein 1 and promotes the
proliferation of leukemic cells (22). In previous years, several genetic
markers in AML have been identified, leading to increased
understanding and providing targets for treatment of the disease
(23–26). However, the relevant aberrations
that contribute to the pathogenesis of this disease remain to be
fully elucidated. In the present study, the importance of S1PR1 in
regulating the apoptosis and proliferation of AML cells were
identified. S1PR1 inhibited AML cell apoptosis through interfering
with mitochondrion-mediated apoptosis processes, ROS generation and
JNK MAPK signaling. S1PR1 also promoted the proliferation of AML
cells, which was at least partially due to the activation of ERK
signaling by S1PR1.
ROS are diffusible chemical mediators. They can act
directly on the apoptotic machinery, by accelerating depolarization
and causing dysfunction of mitochondria (20), which in turn, function as a major
source of ROS in the course of apoptosis (21). The results of the present study
suggested that S1PR1 prevented mitochondrion-associated apoptosis
and ROS generation, and confirmed the importance of S1PR1 as an
anti-apoptotic factor, with elucidation of the corresponding
mechanism. However, the upstream reason for the suppression of
apoptosis remains to be elucidated, in terms of the reciprocal
causation between mitochodria injury and ROS generation.
Impaired apoptosis can promote cancer cell survival.
In the evolutionarily conserved 'stress' pathway-mitochondria
signal transduction of apoptosis, the Bcl-2 family are core cell
death machinery, and consist of pro-apoptotic proteins, including
Bcl-2-associated death promoter and Bax, and anti-apoptotic
proteins, including Bcl-2 and Bcl-extra large, which interact to
maintain a balance (27). When
anti-apoptotic Bcl-2 family members are overexpressed, the ratio of
pro- and anti-apoptotic Bcl-2 family members is disturbed, which in
turn potentially drives malignant progression and confers
chemoresistance to cancer cells (23). According to the observations in the
present study, S1PR1 inhibited mitochondrion-associated apoptosis
by reducing the levels of Bax and preventing the cleavage of
caspase-3 (Fig. 4). Although the
inhibition of ROS generation by S1PR1 may explain the
anti-apoptotic mechanism of the protein, there may be other factors
involved in the S1PR1 knockdown-induced apoptosis of AML cells in
addition to ROS.
MAPK signaling pathways are crucial in cell
proliferation and apoptosis. Until now, three MAPK families have
been clearly characterized: ERK1/2, JNK/MAPK and p38 kinase. The
distinct MAPK members mediate different physiological activities;
the ERKs function in proliferation and differentiation, whereas the
JNKs and p38 MAPKs are predominantly involved in the stress
response and apoptosis through responding to various stresses
(25). Several studies have
suggested that stimulated ERK1 activity results in enhanced cell
proliferation (24,26). Of note, S1PR1 can promote the
MKK1/ERK signaling pathway, which was observed in the AML cells
examined in the present study. Therefore, the enhancement of ERK
activation may be a reason for the S1PR1-mediated promotion of AML
cell proliferation observed in the present study. However, the
mechanism by which S1PR1 enhances MKK/ERK signaling remains to be
elucidated.
According to the results of the present study, the
reduction of ROS is likely to be involved in S1PR1-induced cell
proliferation. Although the S1PR1-induced reduction of ROS levels
may be one reason for the inhibition of JNK signaling by S1PR1, the
possibility that S1PR1 counteracts JNK activation in a
ROS-independent manner cannot be excluded. Further investigations
are required to determine whether S1PR1 suppresses JNK signaling by
other mechanisms.
In conclusion, the present study identified the
anti-apoptotic/pro-proliferative roles of S1PR1 in AML cells, and
elucidated the corresponding mechanism. These findings may provide
insights into the molecular mechanism underlying the progression
and pathogenesis of AML, and benefit the development of
therapeutical approaches for the disease.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant nos. 81100362 and 81570148).
References
|
1
|
Pyne NJ and Pyne S: Sphingosine
1-phosphate and cancer. Nat Rev Cancer. 10:489–503. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee H, Deng J, Kujawski M, Yang C, Liu Y,
Herrmann A, Kortylewski M, Horne D, Somlo G, Forman S, et al:
STAT3-induced S1PR1 expression is crucial for persistent STAT3
activation in tumors. Nat Med. 16:1421–1428. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chae SS, Paik JH, Furneaux H and Hla T:
Requirement for sphingosine 1-phosphate receptor-1 in tumor
angiogenesis demonstrated by in vivo RNA interference. J Clin
Invest. 114:1082–1089. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Deng J, Liu Y, Lee H, Herrmann A, Zhang W,
Zhang C, Shen S, Priceman SJ, Kujawski M, Pal SK, et al:
S1PR1-STAT3 signaling is crucial for myeloid cell colonization at
future metastatic sites. Cancer Cell. 21:642–654. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yoshida Y, Nakada M, Harada T, Tanaka S,
Furuta T, Hayashi Y, Kita D, Uchiyama N, Hayashi Y and Hamada J:
The expression level of sphingosine-1-phosphate receptor type 1 is
related to MIB-1 labeling index and predicts survival of
glioblastoma patients. J Neurooncol. 98:41–47. 2010. View Article : Google Scholar
|
|
6
|
Matsuoka Y, Nagahara Y, Ikekita M and
Shinomiya T: A novel immunosuppressive agent FTY720 induced Akt
dephosphorylation in leukemia cells. Br J Pharmacol. 138:1303–1312.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang JD, Takahara S, Nonomura N, Ichimaru
N, Toki K, Azuma H, Matsumiya K, Okuyama A and Suzuki S: Early
induction of apoptosis in androgen-independent prostate cancer cell
line by FTY720 requires caspase-3 activation. Prostate. 40:50–55.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hung JH, Lu YS, Wang YC, Ma YH, Wang DS,
Kulp SK, Muthusamy N, Byrd JC, Cheng AL and Chen CS: FTY720 induces
apoptosis in hepatocellular carcinoma cells through activation of
protein kinase C delta signaling. Cancer Res. 68:1204–1212. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Adams JM: Ways of dying: Multiple pathways
to apoptosis. Genes Dev. 17:2481–2495. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fiers W, Beyaert R, Declercq W and
Vandenabeele P: More than one way to die: Apoptosis, necrosis and
reactive oxygen damage. Oncogene. 18:7719–7730. 1999. View Article : Google Scholar
|
|
11
|
Simon HU, Haj-Yehia A and Levi-Schaffer F:
Role of reactive oxygen species (ROS) in apoptosis induction.
Apoptosis. 5:415–418. 2000. View Article : Google Scholar
|
|
12
|
Fiers W, Beyaert R, Declercq W and
Vandenabeele P: More than one way to die: Apoptosis, necrosis and
reactive oxygen damage. Oncogene. 18:7719–7730. 1999. View Article : Google Scholar
|
|
13
|
Chen YR, Shrivastava A and Tan TH:
Down-regulation of the c-Jun N-terminal kinase (JNK) phosphatase
M3/6 and activation of JNK by hydrogen peroxide and pyrrolidine
dithiocarbamate. Oncogene. 20:367–374. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sakon S, Xue X, Takekawa M, Sasazuki T,
Okazaki T, Kojima Y, Piao JH, Yagita H, Okumura K, Doi T and Nakano
H: NF-kappaB inhibits TNF-induced accumulation of ROS that mediate
prolonged MAPK activation and necrotic cell death. EMBO J.
22:3898–3909. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kamata H, Honda S, Maeda S, Chang L,
Hirata H and Karin M: Reactive oxygen species promote
TNFalpha-induced death and sustained JNK activation by inhibiting
MAP kinase phosphatases. Cell. 120:649–661. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Grigoryev S, Salzberg A, Berg A,
Harris-Becker A, Loughran T and Claxton D: Genome-wide mapping of
large organized heterochromatin domains reveals hotspots of
epigenetic and transcriptional changes associated with myeloid
differentiation and acute myeloid leukemia (565.1). The FASEB
Journal. 28:565.5612014.
|
|
17
|
Sarkisyan G, Gay LJ, Nguyen N, Felding BH
and Rosen H: Host endothelial S1PR1 regulation of vascular
permeability modulates tumor growth. Am J Physiol Cell Physiol.
307:C14–C24. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hamidi S, Schäfer-Korting M and Weindl G:
TLR2/1 and sphingosine 1-phosphate modulate inflammation,
myofibroblast differentiation and cell migration in fibroblasts.
Biochim Biophys Acta. 1841:484–494. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang T, Chen F, Chen Z, Wu YF, Xu XL,
Zheng S and He X: Honokiol induces apoptosis through
p53-independent pathway in human colorectal cell line RKO. World J
Gastroenterol. 10:2205–2208. 2004.PubMed/NCBI
|
|
20
|
Jabs T: Reactive oxygen intermediates as
mediators of programmed cell death in plants and animals. Biochem
Pharmacol. 57:231–245. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cai J and Jones DP: Mitochondrial redox
signaling during apoptosis. J Bioenerg Biomembr. 31:327–334. 1999.
View Article : Google Scholar
|
|
22
|
Cristóbal I, Blanco FJ, Garcia-Orti L,
Marcotegui N, Vicente C, Rifon J, Novo FJ, Bandres E, Calasanz MJ,
Bernabeu C and Odero MD: SETBP1 overexpression is a novel
leukemogenic mechanism that predicts adverse outcome in elderly
patients with acute myeloid leukemia. Blood. 115:615–625. 2010.
View Article : Google Scholar
|
|
23
|
Adams J and Cory S: The Bcl-2 apoptotic
switch in cancer development and therapy. Oncogene. 26:1324–1337.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pagès G, Lenormand P, L'Allemain G,
Chambard JC, Meloche S and Pouysségur J: Mitogen-activated protein
kinases p42mapk and p44mapk are required for fibroblast
proliferation. Proc Natl Acad Sci USA. 90:8319–8323. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rubinfeld H and Seger R: The ERK cascade:
A prototype of MAPK signaling. Mol Biotechnol. 31:151–174. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Seger R and Krebs EG: The MAPK signaling
cascade. FASEB J. 9:726–735. 1995.PubMed/NCBI
|
|
27
|
Chalah A and Khosravi-Far R: The
mitochondrial death pathway. Adv Exp Med Biol. 615:25–45. 2008.
View Article : Google Scholar : PubMed/NCBI
|