Introduction
Hyperhomocysteinemia, defined as elevated levels of
homocysteine (Hcy) in blood plasma total is identified as an
independent risk factor for the development of atherosclerosis
(1–3). Previous studies have determined that
the elevation of Hcy levels in the blood plasma may lead to
endothelial dysfunction (4,5),
which is identified as an early event in the pathogenesis of
atherosclerosis (6,7). A previous study reported that the
apoptosis of endothelial cells contributed to endothelial
dysfunction and destabilization of atherosclerotic plaques and
thrombosis (8). Apoptosis or
programmed cell death differs from necrosis in that it is an active
process of cell suicide. In the vasculature, misdirected control of
apoptosis in endothelial cells may lead to pathological conditions
including inflammation, clotting and recruitment of smooth muscle
cells. A previous study demonstrated that Hcy may activate a
mitochondrial pathway, which may lead to reduction of the
mitochondrial membrane potential (Δψm) and induce the apoptosis of
endothelial cells and result in cardiac dysfunction in vitro
(9).
Death-associated protein kinase (DAPK), is an
established mediator of programmed cell death (10). It has previously been observed to
upregulate expression levels in atherosclerotic lesions (11). The upregulation of DAPK was
observed to increase cell turnover and arterial wall instability,
which increased susceptibility to low-density lipoprotein
absorption (12). DAPK may
function as a tumor suppressor due to its ability to promote
apoptosis and autophagy (13,14),
suppress cellular transformation (15) and inhibit metastasis (16,17).
Additionally, DAPK may be activated by various stimuli, including
tumor necrosis factor (TNF-α), interferon-γ and p53; therefore,
acts as a converging point for apoptotic signaling (17–19).
DAPK is upstream of the caspases, with the exception of caspase 8,
and may induce caspase-independent cell death or autophagy.
Previous studies have determined that DAPK contributed to shear
stress-induced endothelial apoptosis (20,21).
However, the importance of DAPK in Hcy-induced apoptosis in
endothelial cells remains to be fully elucidated.
The present study investigated the function of DAPK
in Hcy-induced apoptosis in endothelial cells. It was determined
that DAPK may contribute to the modulation of the mitochondrial
pathway in Hcy-induced apoptosis in human umbilical vein
endothelial cells (HUVECs).
Materials and methods
Materials
D,L-Hcy,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
and rhodamine 123 (Rh123), a mitochondrial-specific fluorescent
dye, were purchased from Sigma-Aldrich, Merck Millipore (Darmstadt,
Germany). Dulbecco's modified Eagle's medium (DMEM) and fetal
bovine serum (FBS) were purchased from Gibco; Thermo Fisher
Scientific, Inc. (Waltham, MA, USA). Dimethyl sulfoxide (DMSO) was
purchased from Shenggong Biology Engineering Technology Service,
Ltd. (Shanghai, China). The Bicinchoninic Acid (BCA) protein assay
kit and Annexin V-fluorescein isothiocyanate (FITC) Apoptosis
Detection kit were purchased from KeyGen Biotech Co., Ltd.
(Nanjing, China). Polyclonal antibodies against β-actin (cat. no.
AA128; 1:1,000) and horseradish peroxidase-conjugated secondary
antibodies (goat-anti rabbit; cat. no. A0208; 1:1,000; goat-anti
mouse; cat. no. A0216; 1:2,000) were purchased from Beyotime
Institute of Biotechnology (Shanghai, China). The DAPK antibody
(cat. no. ab109382; 1:1,000) was obtained from Abcam (Cambridge,
UK). Polyclonal antibodies against B cell leukemia/lymphoma 2
(Bcl2; cat. no. 2876; 1:1,000), Bcl2-associated X protein (Bax;
cat. no. 2772; 1:1,000), caspase 3 (cat. no. 9662s; 1:1,000) and
polyclonal ADP-ribose polymerase (PARP; cat. no. 9542; 1:1,000)
were purchased from Cell Signaling Technology, Inc. (Danvers, MA,
USA). The western blotting detection kit was purchased from EMD
Millipore (Billerica, MA, USA).
Cell culture
HUVECs were obtained from China Center for Type
Culture Collection (Wuhan, China) and maintained in DMEM
supplemented with 10% FBS at 37°C in a humidified atmosphere with
5% CO2. The medium was changed every 2–3 days. Endothelial cells of
passage 4–6 in the actively growing condition were used for the
subsequent experiments.
Cell viability assay
Cell viability was evaluated by MTT assays as
previously described (22).
Briefly, non-transfection HUVECs were seeded in 96-well plates
(1.0×104 cells/well) and were treated with Hcy at
concentrations of 0, 0.1, 1, 5, 10 and 15 mM. After 24 h, 500 µg/ml
MTT reagent was added and the cells were incubated for an
additional 4 h. Subsequently, 150 µl DMSO was added to dissolve the
formazan crystals. The absorbance was determined using a microplate
reader (Thermo Fisher Scientific, Inc.) at 570 nm.
Cell transfection
DAPK small interfering RNA (siRNA) and control siRNA
were obtained from GenePharma Co., Ltd. (Shanghai, China). HUVECs
were cultured in 6-well plates at a density of 2.0×105 cells/well
for 24 h and then transfected with DAPK siRNA (30 nM) or control
siRNA (30 nM) in DMEM medium without FBS using Lipofectamine 2000
transfection reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol.
Hoechst 33342 staining for nuclei
fragmentation
HUVECs were treated with 0, 5, 10 mM Hcy for 24 h.
The cells were fixed with 4% paraformaldehyde for 30 min at room
temperature, then stained with Hoechst 33342 (10 µg/ml) at 37°C for
20 min, followed by two washes with phosphate-buffered saline
(PBS). Subsequently, the cells were resuspended with PBS in order
to determine alterations in their nuclear morphology under a
fluorescence microscope (1X71; Olympus Corporation, Tokyo,
Japan).
Apoptosis analysis by flow
cytometry
Flow cytometry was used to detect the apoptotic rate
of HUVECs using the Annexin V-FITC/propidium iodide (PI) staining
kit. Following exposure to different concentrations of Hcy (0, 5
and 10 mM), cells were collected and washed with PBS and
resuspended in binding buffer containing Annexin V-FITC and PI
according to the manufacturer's protocol. Subsequent to staining,
cells were analyzed using a flow cytometer.
Flow cytometry determination of
Δψm
The alterations in Δψm were investigated using Rh123
and flow cytometry. Cells were seeded in 6-well plates
(2.0×105 cells/well), transfected with siRNA, which
downregulated DAPK expression for 48 h and then treated with 10 mM
Hcy for an additional 24 h. Subsequently, cells were incubated with
the Rh123 (10 µM) at 37°C in the dark for 20 min. Following
filtration through 200-mesh sieve with pore size of 75 µm, the
samples were analyzed using a flow cytometer.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Cells were collected to isolate total RNA and remove
genomic DNA using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) in a sterile, RNase-free environment. Reverse
transcription was performed using RevertAid Reverse Transcriptase
(Thermo Fisher Scientific, Inc.) at 37°C for 1 h. The qPCR reaction
was performed using Maxima SYBR Green PCR Master mix (Thermo Fisher
Scientific, Inc.). The 25 µl final reaction volume was comprised of
12.5 µl Maxima SYBR Green PCR Master mix, 0.3 µM primer and 1 µl
cDNA. The thermocycling conditions were as follows: Initial step of
50°C for 2 min, followed by a second step at 95°C for 15 min, then
40 cycles of 95°C for 15 sec and 60°C for 60 sec. Fold changes in
target gene expression between treatments and controls were
determined using the 2−ΔΔCq method (23), normalizing to GAPDH RNA expression
as an internal reference. All results were repeated in six
independent experiments and performed in triplicate each time. The
following primers for qPCR were used: DAPK forward,
ACACATTGCCCTTCATCTGG and reverse AGTATTGCCGTGCCTGTC TT; GAPDH
forward CGCTCTCTGCTCCTCCTGTTC and reverse
ATCCGTTGACTCCGACCTTCAC.
Protein analysis
For protein analysis, cells were collected following
each experiment and lysed with radioimmunoprecipitation assay
buffer and ultrasound on ice. The supernatant fluids were collected
following centrifugation at 13,000 × g for 5 min at 4°C. BCA
Protein Assay kit was used to determine the protein concentrations.
Gel electrophoresis was performed using 10% sodium dodecyl
sulphate-polyacrylamide gel and transferred to 0.45 µm
polyvinylidene fluoride membranes (GE Healthcare Life Sciences,
Chalfont, UK). The membranes were immersed in 5% non-fat milk
blocking buffer for 1 h. The membranes were incubated overnight at
4°C with the primary antibodies (1:1,000), followed by appropriate
horseradish peroxidase (HRP)-conjugated secondary antibodies
(1:1,000) at room temperature for 1 h and Immobilon Western
Chemiluminescent HRP substrate (EMD Millipore). Gel-Pro Analyzer
version 6.0 (Media Cybernetics, Inc., Rockville, MD, USA) was used
to extract quantitative information from the electrophoretic
gels.
Statistical analysis
Data are expressed as the mean ± standard deviation
from at least three different experiments. Comparisons between
groups were performed using one-way analysis of variance followed
by Dunnett's test using using SPSS version 13.0 (SPSS, Inc.,
Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Hcy reduces the survival of HUVECs and
induces apoptosis
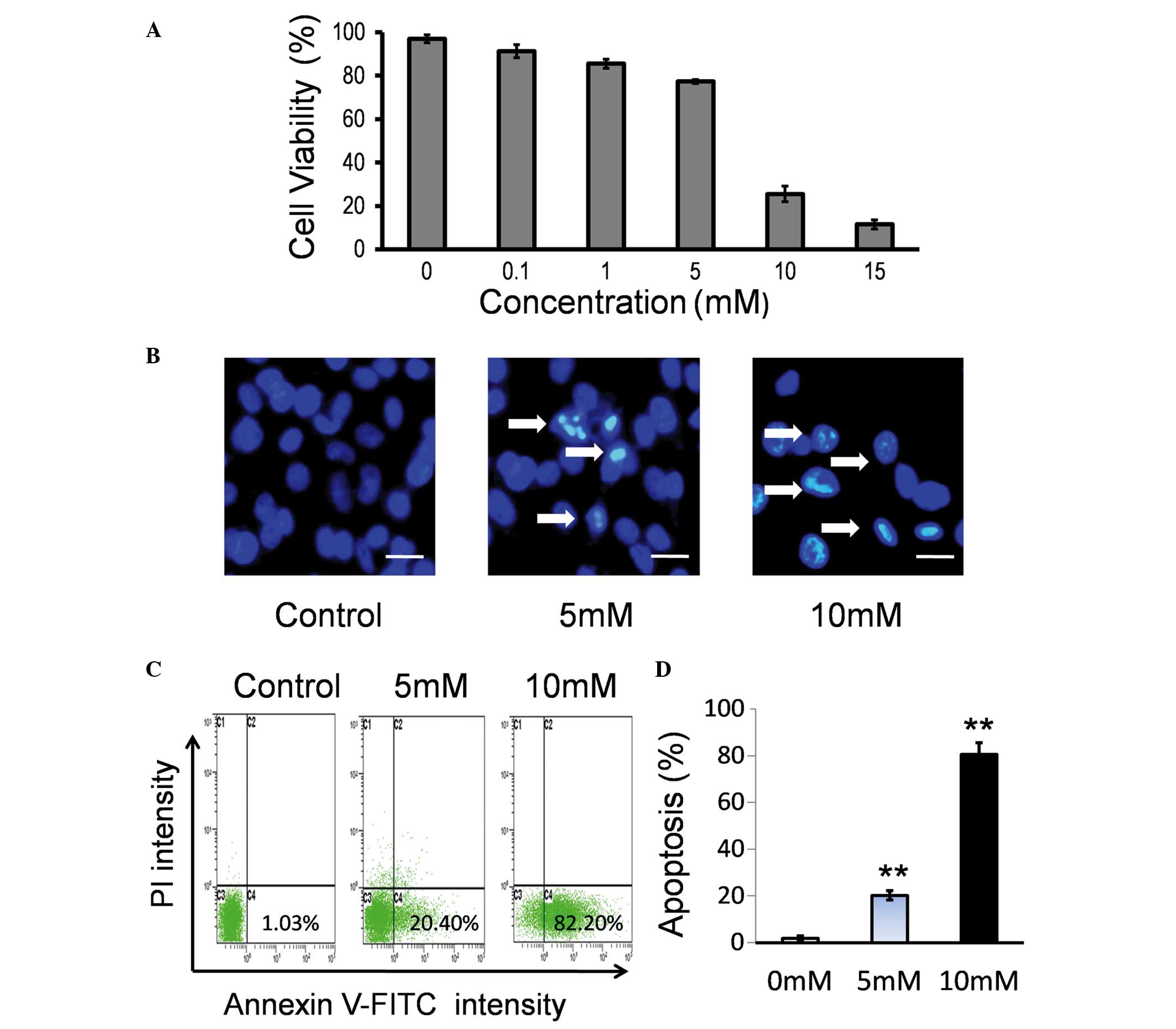
To examine the effect of Hcy on the viability of
endothelial cells, HUVECs were treated with various concentrations
(0.1–15 mM) of Hcy. No significant difference was identified
between 0.1 mM Hcy and the control group (P>0.05; Fig. 1A). Hcy at 1, 5, 10 and 15 mM
significantly inhibited cell viability in a dose-dependent manner
compared with the control group (P<0.05; Fig. 1A).
The major features of apoptotic cell death are DNA
fragmentation and loss of the asymmetry of the plasma membrane. To
test the effect of Hcy on cell viability, morphological changes in
the nuclei were observed using Hoechst 33342 staining and
fluorescence microscopy. As presented in Fig. 1B, Hcy treatment induced nuclear
morphological alterations in the HUVECs, including nuclear
shrinkage and DNA fragmentation. Apoptosis induced by Hcy was
additionally confirmed by Annexin V-FITC/PI staining. HUVECs were
treated with Hcy for 24 h and the percentages of cells undergoing
apoptosis/necrosis were determined by flow cytometry. It was
identified that Hcy may induce cell apoptosis in a dose-dependent
manner (Fig. 1C). A significant
increase in early apoptosis of the Hcy 5 mM and 10 mM treatment
groups was observed compared with the control group (P<0.001;
Fig. 1D).
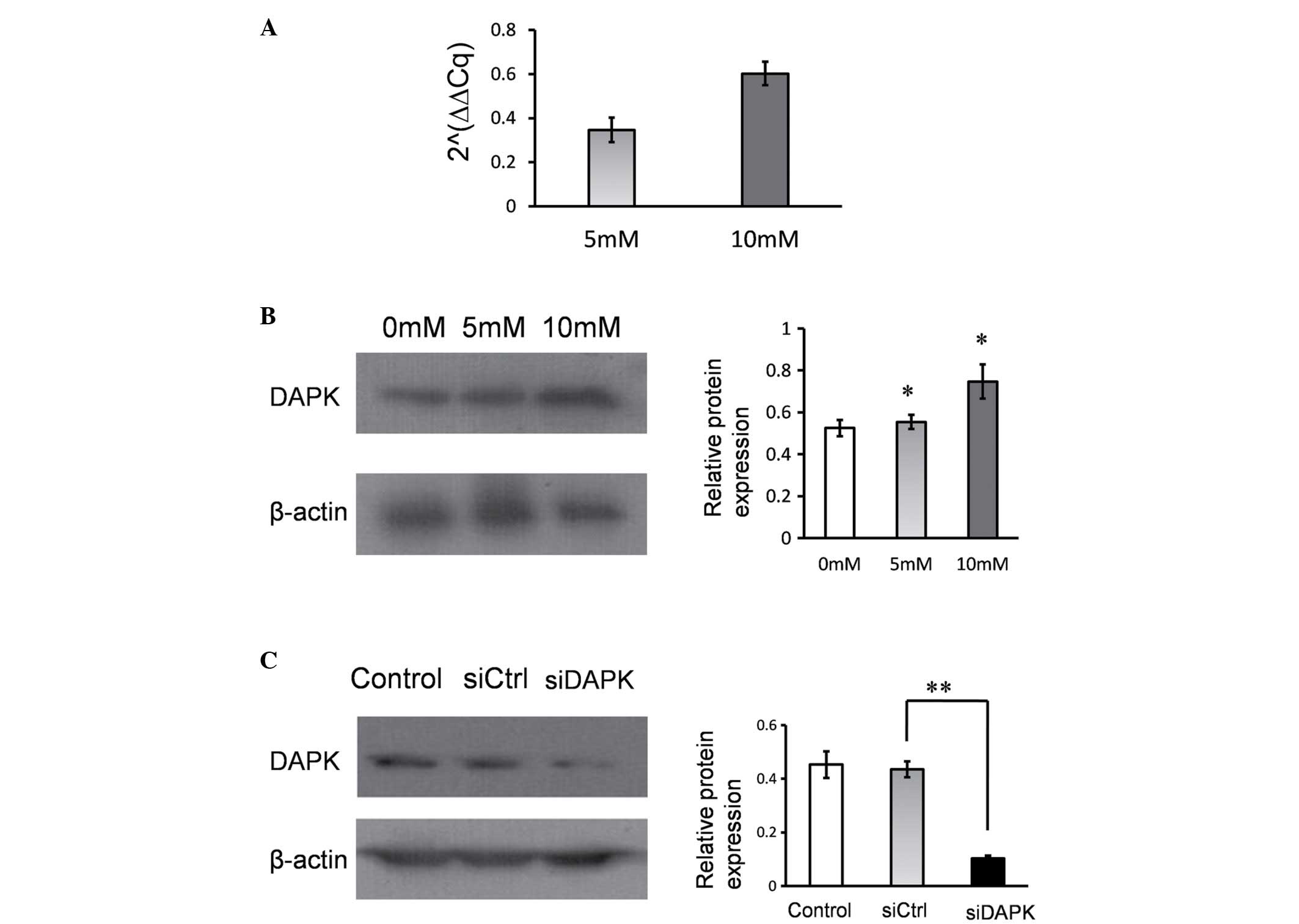
Hcy increases DAPK expression in HUVECs. Following
treatment with Hcy, the mRNA and protein expression levels of DAPK
were determined using RT-qPCR and western blotting. It was
identified that the mRNA and protein levels of DAPK were
significantly upregulated in response to the 5 mM and 10 mM Hcy
treatment groups, when compared with the control (P<0.05;
Fig. 2A and B). Hcy treatment
increased DAPK mRNA and protein expression levels in HUVECs.
DAPK knockdown reduced the Hcy-induced
apoptosis
To determine the effect of DAPK expression on
Hcy-induced apoptosis, DAPK expression was inhibited using siRNA.
DAPK expression was significantly suppressed in HUVECs compared
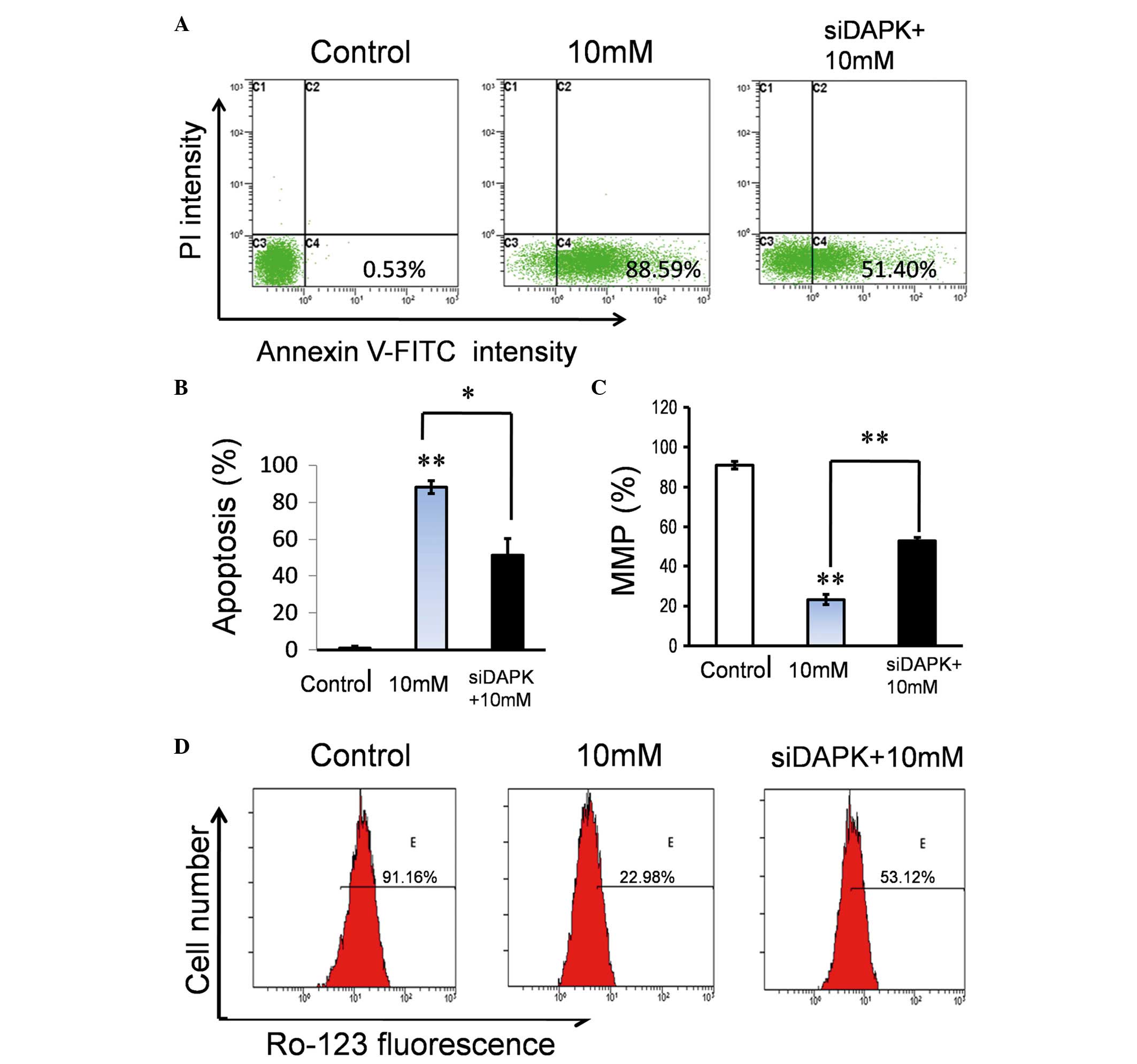
with the non-specific control siRNA group (P<0.001; Fig. 2C). Following treatment with Hcy,
the apoptotic rate of DAPK siRNA-transfected HUVECs was
significantly reduced from 88.59 to 51.40%, compared with the
control group (transfected with non-specific siRNA) (P<0.05;
Fig. 3A and B). These findings
provide additional evidence that DAPK is important for the
mediation of endothelial apoptosis induced by Hcy. Additionally,
cell viability was significantly greater in the siDAPK group
compared with the 10 mM Hcy group (P<0.001; Fig. 3C)
DAPK knockdown attenuates the effect
of Hcy on Δψm
Hcy endothelial cells was associated with DAPK. In
order to determine whether reduction of DAPK expression levels may
alleviate Hcy-induced mitochondrial dysfunction, DAPK
siRNA-transfected cells were treated with Hcy and the Δψm was
evaluated. Following treatment with Hcy, an increase of Δψm was
observed in the DAPK knockdown group from 22.98 to 53.12% (Fig. 3D). This indicated that DAPK was
associated with the Hcy-induced mitochondrial dysfunction.
Knockdown of DAPK attenuates the
effect of Hcy on the expression levels of apoptosis regulators
The data indicated that DAPK was involved in
Hcy-induced apoptosis and with Δψm disruption in HUVECs. The Bcl2
protein family, a large family of apoptosis regulating proteins,
modulated the mitochondrial pathway investigated in the present
study. In order to characterize the function of DAPK in Hcy-induced
apoptosis, the impact of reduced DAPK expression levels via siRNA
transfection on Bcl2 family proteins was investigated in HUVECs
treated with Hcy using western blot analysis (Fig. 4). As presented in Fig. 4A and C, Hcy treatment significantly
reduced the ratio of Bcl2 to Bax (P<0.05), whereas the knockdown
of DAPK significantly reversed the effect of Hcy treatment on Bax
and Bcl2 levels (P<0.05). In addition, caspase 3 and PARP
activation was examined. Hcy treatment resulted in significantly
increased caspase 3 cleavage and reduced PARP expression levels in
HUVECs (P<0.01; Fig. 4C).
Caspase 3 cleavage, characterized by the appearance of 17 and 19
kDa protein band and PARP cleavage characterized by a 89 kDa
protein band reduced in cells transfected with siDAPK compared with
the cells transfected with non-specific siRNA. Cleaved caspase 3
expression levels were significantly reduced in the siDAPK group
compared with the 10 mM Hcy treatment group (P<0.05; Fig. 4C).
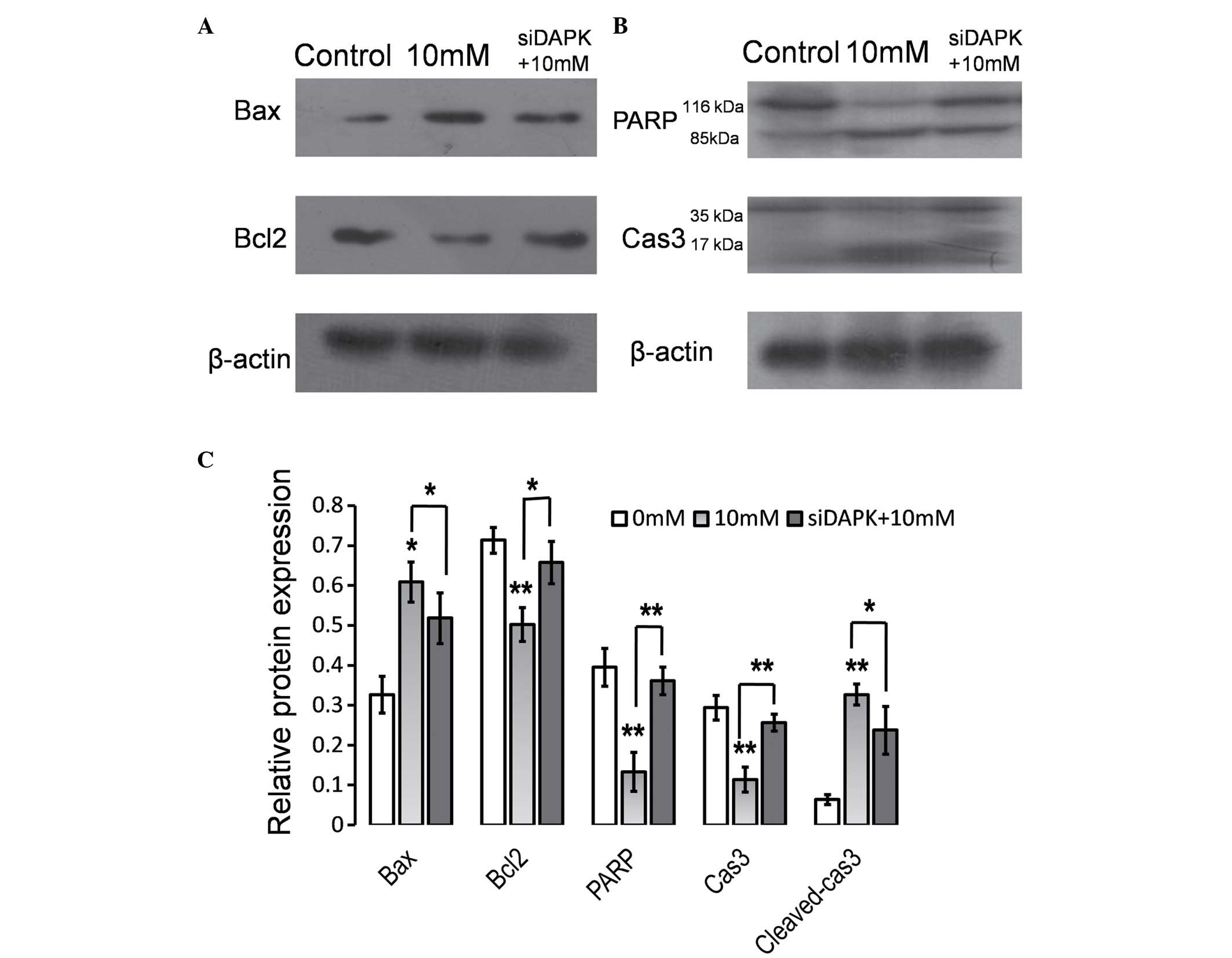 | Figure 4.Suppression of DAPK attenuated the
effect of Hcy on the expression levels of apoptosis regulators.
HUVECs were transfected with DAPK-specific siRNA and 48 h
subsequent to transfection, cells were treated with Hcy (10 mM) for
24 h. Whole-cell extracts were prepared and probed for (A) Bcl2,
Bax, (B) PARP and Cas3 by western blot analysis. (C) Quantification
of the western blotting. Data are expressed as the mean ± standard
deviation of three independent experiments. *P<0.05 and
**P<0.01 vs. control. DAPK, death-associated protein kinase;
Hcy, homocysteine; HUVECS, human umbilical vein endothelial cells;
siRNA, small interfering RNA; Bcl2, B cell leukemia/lymphoma 2;
Bax, Bcl2-associated X protein; PARP, poly ADP-ribose polymerase;
Cas3, caspase 3. |
Discussion
The present study investigated the importance of
DAPK for the possible mechanism that triggers Hcy-induced apoptosis
in HUVECs. Initially, it was confirmed that Hcy reduced the
viability of HUVECs and induced apoptosis. In addition, it was
determined that Hcy upregulated DAPK expression in a dose-dependent
manner. Additionally, it was revealed that inhibition of DAPK may
attenuate apoptosis and dissipation of the Δψm. The underlying
molecular mechanisms were also investigated and it was determined
that DAPK participated in Hcy-induced apoptosis by reducing the
ratio of Bcl2/Bax and activation of caspase 3 in HUVECs.
Increased levels of Hcy in blood plasma have been
identified as an independent risk factor for the development of
atherosclerosis (1–3). In atherosclerosis, increased
apoptosis has been established to contribute to prolonged
inflammatory response, plaque instability, rupture and thrombus
formations (11). Previous studies
have also reported apoptosis of endothelial cells associated with
Hcy (24–27). The results of the present study are
in agreement with this, as it was evident that Hcy reduced cell
viability and induced apoptosis in a dose-dependent manner.
DAPK is an important protein kinase for the
modulation of apoptotic pathways (28). A previous study identified that
DAPK functioned as a positive mediator of apoptosis induced by
various stimuli, including TNF-α, interferon-γ and p53 (29). It was previously reported that DAPK
activity may be involved in TNF-α-induced apoptotic pathways in
bovine aortic endothelial cells (20). To the best of our knowledge, the
present study is the first to reported that Hcy treatment increased
mRNA and protein DAPK expression levels. This suggested that DAPK
may contribute to the apoptotic effect of Hcy in endothelial cells.
When inhibition of DAPK was induced, a reduction in Hcy-induced
apoptosis was observed in endothelial cells, indicating that DAPK
is crucial for the mediation of Hcy-induced apoptosis.
A previous study determined that Δψm loss, an early
occurring event, may be directly associated with apoptosis
(30). As Δψm is reduced,
mitochondrial permeability transition pores are opened and in turn
release cytochrome c and other pro-apoptotic molecules from
the intermembrane space into the cytosol. It has been previously
demonstrated that Hcy activates the mitochondrial pathway leading
to a reduction in Δψm and apoptosis in cardiac microvascular
endothelial cells (26). The
current study determined that Hcy treatment resulted in a reduction
of Δψm in HUVECs, which is consistent with previous studies
(26,31). It is of note, that the reduction of
Δψm may be attenuated by the knockdown of DAPK, as it has been
identified to be important for Hcy-induced mitochondrial
dysfunction. Therefore, this may be the possible mechanism behind
endothelial apoptosis induced by Hcy.
Bcl2 family proteins are involved in the
mitochondria-dependent apoptosis pathway, which includes
anti-apoptotic proteins and pro-apoptotic proteins such as Bcl2 and
Bax (32). The present study
demonstrated that following Hcy treatment the protein expression
levels of Bax were increased, whereas the expression levels of Bcl2
were reduced. It is of note that downregulation of DAPK expression
reversed this response. Caspase activation is one of the processes
that signify the onset of apoptosis, and caspase 3 has been
considered to be a central component of the proteolytic cascade
during apoptosis, as it may cleave various nuclear proteins,
including PARP, which may lead to atypical apoptotic DNA
fragmentation (33). The present
study demonstrated that exposure to Hcy activated caspase 3,
whereas the knockdown of DAPK reduced this activation. Therefore,
it is possible that Hcy-induced apoptosis in HUVECs is associated
with modulation of the Bcl2/Bax ratio and activation of caspase-3
by DAPK.
Acknowledgements
The present study was supported by the Wu Jieping
Medical Foundation (grant no. 320.6750.12265).
References
|
1
|
Guilland JC, Favier A, de Courcy G Potier,
Galan P and Hercberg S: Hyperhomocysteinemia: An independent risk
factor or a simple marker of vascular disease? 1. Basic data.
Pathol Biol (Paris). 51:101–110. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
de Koning AB Lawrence, Werstuck GH, Zhou J
and Austin RC: Hyperhomocysteinemia and its role in the development
of atherosclerosis. Clin Biochem. 36:431–441. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Clarke R, Daly L, Robinson K, Naughten E,
Cahalane S, Fowler B and Graham I: Hyperhomocysteinemia: An
independent risk factor for vascular disease. N Engl J Med.
324:1149–1155. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Horvath B, Szapary L, Debreceni L, Feher
G, Kenyeres P, Fulop A, Battyani I and Toth K: Effect of Sclerovit
on endothelial dysfunction, hemorheological parameters, platelet
aggregation, plasma concentration of homocysteine and progression
of atherosclerosis in patients with vascular diseases. Clin
Hemorheol Microcirc. 42:19–28. 2009.PubMed/NCBI
|
|
5
|
Briasoulis A, Tousoulis D, Androulakis ES,
Papageorgiou N, Latsios G and Stefanadis C: Endothelial dysfunction
and atherosclerosis: Focus on novel therapeutic approaches. Recent
Pat Cardiovasc Drug Discov. 7:21–32. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Davignon J and Ganz P: Role of endothelial
dysfunction in atherosclerosis. Circulation. 109(23): Suppl 1.
III27–III32. 2004.PubMed/NCBI
|
|
7
|
Martin BJ and Anderson TJ: Risk prediction
in cardiovascular disease: The prognostic significance of
endothelial dysfunction. Can J Cardiol. 25:(Suppl A). A15–A20.
2009. View Article : Google Scholar
|
|
8
|
Chen F, Eriksson P, Kimura T, Herzfeld I
and Valen G: Apoptosis and angiogenesis are induced in the unstable
coronary atherosclerotic plaque. Coron Artery Dis. 16:191–197.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kumar D and Jugdutt BI: Apoptosis and
oxidants in the heart. J Lab Clin Med. 142:288–297. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cohen O and Kimchi A: DAP-kinase: From
functional gene cloning to establishment of its role in apoptosis
and cancer. Cell Death Differ. 8:6–15. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Martinet W, Schrijvers DM, De Meyer GR,
Thielemans J, Knaapen MW, Herman AG and Kockx MM: Gene expression
profiling of apoptosis-related genes in human atherosclerosis:
Upregulation of death-associated protein kinase. Arterioscler
Thromb Vasc Biol. 22:2023–2029. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schumacher AM, Velentza AV and Watterson
DM: Death-associated protein kinase as a potential therapeutic
target. Expert Opin Ther Targets. 6:497–506. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Inbal B, Bialik S, Sabanay I, Shani G and
Kimchi A: DAP kinase and DRP-1 mediate membrane blebbing and the
formation of autophagic vesicles during programmed cell death. J
Cell Biol. 157:455–468. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bialik S and Kimchi A: The
death-associated protein kinases: Structure, function, and beyond.
Annu Rev Biochem. 75:189–210. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Raveh T, Droguett G, Horwitz MS, DePinho
RA and Kimchi A: DAP kinase activates a p19ARF/p53-mediated
apoptotic checkpoint to suppress oncogenic transformation. Nat Cell
Biol. 3:1–7. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Michie AM, McCaig AM, Nakagawa R and
Vukovic M: Death-associated protein kinase (DAPK) and signal
transduction: Regulation in cancer. FEBS J. 277:74–80. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen RH, Wang WJ and Kuo JC: The tumor
suppressor DAP-kinase links cell adhesion and cytoskeleton
reorganization to cell death regulation. J Biomed Sci. 13:193–199.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cohen O, Inbal B, Kissil JL, Raveh T,
Berissi H, Spivak-Kroizaman T, Feinstein E and Kimchi A: DAP-kinase
participates in TNF-alpha- and Fas-induced apoptosis and its
function requires the death domain. J Cell Biol. 146:141–148. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pelled D, Raveh T, Riebeling C, Fridkin M,
Berissi H, Futerman AH and Kimchi A: Death-associated protein (DAP)
kinase plays a central role in ceramide-induced apoptosis in
cultured hippocampal neurons. J Biol Chem. 277:1957–1961. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rennier K and Ji JY: Shear stress
regulates expression of death-associated protein kinase in
suppressing TNFα-induced endothelial apoptosis. J Cell Physiol.
227:2398–2411. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rennier K and Ji JY: Effect of shear
stress and substrate on endothelial DAPK expression, caspase
activity, and apoptosis. BMC Res Notes. 6:102013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hua P, Sun M, Zhang G, Zhang Y, Tian X, Li
X, Cui R and Zhang X: Cepharanthine induces apoptosis through
reactive oxygen species and mitochondrial dysfunction in human
non-small-cell lung cancer cells. Biochem Biophys Res Commun.
460:136–142. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Suhara T, Fukuo K, Yasuda O, Tsubakimoto
M, Takemura Y, Kawamoto H, Yokoi T, Mogi M, Kaimoto T and Ogihara
T: Homocysteine enhances endothelial apoptosis via upregulation of
Fas-mediated pathways. Hypertension. 43:1208–1213. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang C, Cai Y, Adachi MT, Oshiro S, Aso
T, Kaufman RJ and Kitajima S: Homocysteine induces programmed cell
death in human vascular endothelial cells through activation of the
unfolded protein response. J Biol Chem. 276:35867–35874. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tyagi N, Ovechkin AV, Lominadze D, Moshal
KS and Tyagi SC: Mitochondrial mechanism of microvascular
endothelial cells apoptosis in hyperhomocysteinemia. J Cell
Biochem. 98:1150–1162. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sipkens JA, Hahn N, van den Brand CS,
Meischl C, Cillessen SA, Smith DE, Juffermans LJ, Musters RJ, Roos
D, Jakobs C, et al: Homocysteine-induced apoptosis in endothelial
cells coincides with nuclear NOX2 and peri-nuclear NOX4 activity.
Cell Biochem Biophys. 67:341–352. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kimchi A: DAP kinase and DAP-3: Novel
positive mediators of apoptosis. Ann Rheum Dis. 58:(Suppl 1).
I14–I19. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yoo HJ, Byun HJ, Kim BR, Lee KH, Park SY
and Rho SB: DAPk1 inhibits NF-κB activation through TNF-α and
INF-γ-induced apoptosis. Cell Signal. 24:1471–1477. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lemasters JJ: V. Necrapoptosis and the
mitochondrial permeability transition: Shared pathways to necrosis
and apoptosis. Am J Physiol. 276:G1–G6. 1999.PubMed/NCBI
|
|
31
|
Dong D, Wang B, Yin W, Ding X, Yu J and
Kang YJ: Disturbance of copper homeostasis is a mechanism for
homocysteine-induced vascular endothelial cell injury. PloS One.
8:e762092013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
King KL and Cidlowski JA: Cell cycle
regulation and apoptosis. Annu Rev Physiol. 60:601–617. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Porter AG and Jänicke RU: Emerging roles
of caspase-3 in apoptosis. Cell Death Differ. 6:99–104. 1999.
View Article : Google Scholar : PubMed/NCBI
|