Introduction
Glioblastoma multiforme is an aggressive malignant
brain tumor. Patient prognosis is poor, with a median survival of
6–12 months, as the standard treatment is ineffective. A primary
cause of this resistance to treatment is the stem cells of the
cancer cell population, which have unique signaling and
morphogenetic properties (1–3).
Cancer stem cells (CSCs) represent a specific group
of tumor cells that have maximum independence from external
signals. Relatively small numbers of these cells are sufficient for
the rapid activation of invasive growth and metastasis. These cells
are multipotent, capable of constant self-renewal and have the
greatest proliferative activity of all glioblastoma cells (4,5).
Specific features of CSCs include a hypoxic metabolism and an
ability to restore damaged DNA (6,7).
Previously, CSCs were considered to develop from
differentiated cells. However, normal stem cells are now being
viewed as the primary source of CSCs. The modern concept of
carcinogenesis defines cancer and other malignant tumors as a
genetic disease developing in the nucleus of a normal stem cell
that renders its descendants (CSCs) unrecognizable to the immune
system and, thus, safe from elimination (8). The eradication of CSCs is therefore
crucial for cancer treatment; however, currently there are no
effective methods to do this.
One approach to solving this problem is to develop
chemotherapeutic agents that would seek out and destroy these
cells. However, CSCs are a heterogeneous cell population and may
adapt to sublethal exposure by producing new clones with
greater resistance. It has been suggested that CSCs represent a
specific survival mechanism of eukaryotic cells and are the result
of a constant struggle for existence (9–11).
Destruction of this target requires a highly accurate and powerful
tool that exceeds the ability of CSCs to adapt.
Patient stem cells may potentially be this tool. The
ability of stem cells (SCs) to migrate to the tumor node and
interact with cancer cells has been proven (12,13).
Certain treatment strategies, including targeted drug delivery
(14) and metallic nanoparticles
for drug delivery (15), are based
on the migration potential of SCs. In addition, SCs that secretes
specific antibodies within the tumor have been demonstrated to
improve survival in a mouse model (16). However, these strategies do not
target the CSCs themselves. This is due to a lack of knowledge
regarding which cancer cells become the target of stem cell
migration, the role of this phenomenon in carcinogenesis and what
stem cell lines should be used to develop antitumor cell therapy.
The answers to these questions will define the direction of future
investigations.
The aim of the present study was to evaluate the
ability of glioblastoma cells to attract various tissue-specific
human stem cells, and to compare normal and cancer stem cells.
Materials and methods
Cell culture
The U251 human glioblastoma cell line (cat. no.
09063001; Sigma-Aldrich; Merck Millipore, Darmstadt, Germany), the
U87 human glioblastoma cell line (ATCC no. HTB-14™; ATCC, Manassas,
VA, USA), the MCF7 human breast cancer cell line (АТСС no.
HTB-22™), the A549 human lung cancer cell line (АТСС no.CCL-185™),
human fibroblasts (ATCC no. PCS-420-013™) and the C6 rat glioma
cell line (ATCC no. CCL-10™) were employed for the purposes of the
present study.
Cell lines were cultured at 37°C with 5%
CO2 in Dulbecco's modified Eagle's medium (DMEM; cat.
no. 61965-026) containing 10% fetal bovine serum (FBS; cat. no.
1347559) and 100Х Antibiotic-Antimycotic (cat. no. 160175; all from
Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The
culture medium was changed every 72 h. The cells for the experiment
were treated with TrypLE Express according to the manufacturer's
instructions (cat. no. 1606073; Gibco; Thermo Fisher Scientific,
Inc.) for 7 min at 37°С and centrifuged for 3 min at 120 × g
at 20°C. The supernatant was subsequently removed and fresh DMEM
was added. The cells were counted in a hemocytometer, following
staining with 0.4% trypan blue (cat. no. 15250061; Gibco; Thermo
Fisher Scientific, Inc.) to assess viability.
To extract CSCs from U87 and U251 human glioblastoma
cell lines, the cells were suspended in dispase/collagenase
solution (dispase, 0.8 U/ml; collagenase, 0.1 U/ml; Roche Applied
Science, Penzberg, Germany) in phosphate-buffered saline (PBS) for
1 h at 37°С. Enzymatic reactions were inactivated in PBS + 5% FBS
and the cells were centrifuged for 5 min at 800 × g at 20°C.
Cells were resuspended in DMEM/F-12 (cat. no. 12634-010; Gibco;
Thermo Fisher Scientific, Inc.) containing L-glutamine, 20 ml/lB-27
supplement (cat. no. 17504044; Gibco; Thermo Fisher Scientific,
Inc.), 20 ng/ml fibroblast growth factor β (FGF-β; cat. no.
PHG0023; Gibco; Thermo Fisher Scientific, Inc.), 20 ng/ml epidermal
growth factor (EGF; cat. no. PHG0311; Gibco; Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin/streptomycin and 5 µg/ml
heparin. Cells were cultured in T75 flasks at 37°C with 5%
CO2. Fresh growth factors were added every 72 h.
Adherent cells were cultured until 80% confluence was reached,
before they were subcultured at a 1:3 ratio. Cluster of
differentiation (CD)133+ cells were selected via
magnetic-activated cell sorting (MACS) using an autoMACS
Pro® and magnetic beads bound to immobilized CD133
antibodies (MiltenyiBiotec, Inc., San Diego, CA, USA), according to
the manufacturer's instructions. The purity was assessed using a
flow cytometer (BD Accuri C6; BD Biosciences, Franklin Lakes, NJ,
USA).
Human neural stem cells (NSCs) derived from the
olfactory epithelium of the superior turbinate were supplied by the
NeuroVita Clinic of Interventional and Restorative Neurology and
Therapy (Moscow, Russia). NSCs were treated with
dispase/collagenase solution and resuspended in growth
factor-containing DMEM/F-12 as described above. Cells were cultured
until cytospheres formed and were then characterized by the
expression of nestin (cat. no. ab22035; Abcam, Cambridge, MA, USA),
Thy1 (CD90; cat. no. ab133350; Abcam); neurofilament 200 (NF200;
cat. no. ab134306; Abcam) and glial fibrillary acidic protein
(GFAP; cat. no. ab7260; Abcam) according to the manufacturer's
recommendations. Cells were fixed with 4% paraformaldehyde
(Sigma-Aldrich; Merck Millipore), washed with PBS, and incubated
overnight at 4°C with 1–5 µg/ml primary antibodies. The cells were
washed in PBS three times, and incubated for 1 h at 37°C with goat
anti-mouseAlexaFluor633-conjugated IgG (cat. no. A-21146;
Invitrogen; Thermo Fisher Scientific, Inc.) or goat anti-rabbit
Alexa Fluor 488-conjugated IgG (cat. no. A-11008; Invitrogen;
Thermo Fisher Scientific, Inc.) secondary antibodies at a
concentration of 5 µg/ml. The cell nuclei were then stained with
DAPI (Sigma-Aldrich; Merck Millipore) before the cells were placed
in 50% glycerin (Sigma-Aldrich; Merck Millipore) and covered with a
0.17-mm cover glass (Menzel-Gläzer, Braunschweig, Germany).
CD133+ NSCs were sorted by MACS using an AutoMACS
Pro®Separator and magnetic beads bound to immobilized
CD133 antibodies (MiltenyiBiotec, Inc.), according to the
manufacturer's instructions. The purity was assessed using a flow
cytometer (BD Accuri C6; BD Biosciences).
Multipotent hematopoietic stem cells (HSCs) were
supplied by the NeuroVita Clinic of Interventional and Restorative
Neurology and Therapy (Moscow, Russia). The cells were obtained
from bone marrow using Pittenger's method (17). Cells was assessed by flow cytometry
based on the presence or absence of the following surface antigens:
CD29+ (cat. no. 130-101-255), CD44+ (cat. no. 130-110-393),
CD73+ (cat. no. 130-103-065), CD90+ (cat. no.
130-097-930) and СD34+ (cat. no. 130-098-142). The
fluorescein isothiocyanate-conjugated antibodies (MiltenyiBiotec,
Inc.) were added to the suspension of studied cells
(1×105) in the concentration recommended by the
manufacturer, incubated for 30 min at 37°C and analyzed using a
flow cytofluorometer (MiltenyiBiotec, Inc.).
Fibroblasts were cultured in DMEM supplemented with
10% FBS, 10 ng/ml EGF, 10 ng/ml FGF-β and 100Х
Antibiotic/Antimycotic, replenished every four days, until
confluence reached 70%. Cells were harvested using 0.25% trypsin
solution for 5 min at 37°С, and centrifuged at 120 × g for 3
min at 20°C. The cells were counted in a hemocytometer, following
staining with 0.4% trypan blue to assess viability. Prior to being
used in the experiment, the material underwent fluorescent staining
using Cell Tracker Red™ CMTPX dye (cat. no. C34552; Molecular
Probes; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions.
Rat HSCs were supplied by the NeuroVita Clinic of
Interventional and Restorative Neurology and Therapy (Moscow,
Russia).
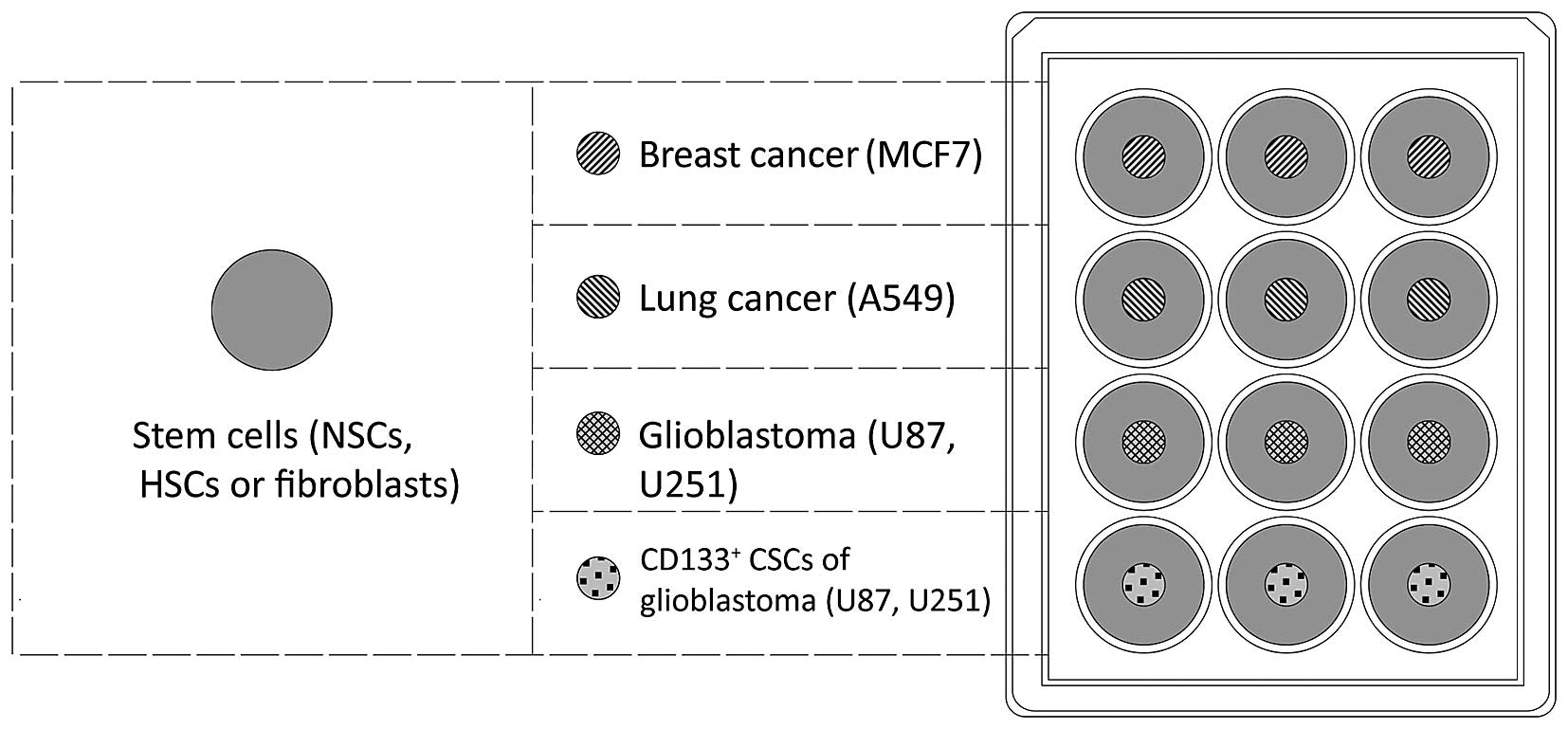
In order to assess the migration of human stem cells
towards malignant cancer cells in vitro, tumor cells
(0.5×104) were seeded onto culture plate inserts (cat.
no. 30012; SPL Life Sciences, Pocheon, Korea) in a 12-well plate
(Fig. 1). NSCs, HSCs and
fibroblasts were stained with the fluorescent Cell Tracker™ Red
CMTPX Dye (Molecular Probes; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions, and added to the
wells (0.5×104 cells/well). The number of cells that
migrated to the walls of culture plate inserts was evaluated using
the monitoring system Cell IQ® (CM Technologies Oy,
Tampere, Finland) following 120 h of co-culture.
Proteome mapping of stem cells
Glioblastoma CSCs consisting of U87 CD133+ NSCs and
HSCs, were lysed using a Mammalian Cell Lysis kit (Sigma-Aldrich;
Merck Millipore) according to the manufacturer's instructions.
Briefly, cells were collected transferred to a conical test tube
and centrifuged at 25°C for 5 min at 420 × g. The
supernatant was decanted and the cells were washed twice by
resuspending the cell pellets in PBS and centrifuging at 420 ×
g for 5 min at 25°C. After decanting the supernatant, cells
were resuspended in cell lysis buffer
(106-107 cells/ml) and incubated for 15 min
on an orbital shaker. The cells were subsequently centrifuged at
12,000 × g for 10 min at 25°C to pellet the cellular debris.
The protein-containing supernatant was removed and transferred to a
fresh test tube. Samples that were used immediately were incubated
on ice, while the remaining samples were stored at −20°C.
Purification from low-molecular weight
components
The subsequent tumor cell lysates were purified from
low-molecular components using Agilent 5K MWCO 4 ml Spin
Concentrators for Proteins (Agilent Technologies GmbH, Waldbronn,
Germany). Samples were placed in a CentriVap vacuum concentrator
(Labconco, Kansas City, MO, USA) and centrifuged at 2000 × g
at 60°C until 200 µl of fluid remained. Water (4 ml) was added and
the samples were centrifuged again under the same conditions. The
samples were then washed three times, leaving 200 µl of the sample.
The sample was washed from the concentrator twice using 200 µl
water. The resultant 600 µl sample was used for downstream
experiments, and total protein was measured using a Bradford
protein assay kit (Sigma-Aldrich; Merck Millipore) according to the
manufacturer's instructions. The volume of lysate containing 300 µg
protein was calculated and dried at 60°C in a CentriVap vacuum
evaporator.
Tryptic cleavage
Trypsinolysis was performed using 150 µl protein
samples. Dried lysates were added to 25 µl 2,2,2-trifluoroethanol
(TFE), 25 µl NH4HCO3 (100 mM) diluted in
water and 2 µl fresh trichloroethylphosphate (TCEP; 50 mM) diluted
in water. The reaction mixture was exposed for 1 h at 60°C, and
then cooled to 25°C. A total of 1 µl fresh aqueous iodoacetamide
(IAA; 84 mM) was added to the sample, which was then exposed for 30
min at 25°C. A total of 100 µl NH4HCO3 (100
mM) solution, 300 µl water and trypsin solution in 1 mM of HCl
(trypsin concentration 100 ng/µl, trypsin: protein ratio, 1:50 by
weight) was subsequently added to the sample, which was then
exposed for 18 h at 37°C. The solution (3 µl) was analyzed using an
LTQOrbitrap XL mass spectrometer (Thermo Fisher Scientific, Inc.)
for the completion of trypsinolysis as described below. Completion
of trypsinolysis was determined by analyzing the tryptic peptide
peaks and by calculating the area under the peaks at m/z 842.51 Da
and 421.76 Da. When the reaction was completed, the content of the
tube was dried at 60°C in the CentriVap.
Liquid chromatography (LC)-mass
spectrometry (MS)/MS analysis using the LTQ Orbitrap XL
Peptides were analyzed using an LTQ Orbitrap XL
instrument (Thermo Fisher Scientific, Inc.) with a NanoSpray
ionization (NSI) ion source coupled to an Ultimate 3000 Dionex
nanoflow LC system (Dionex, Sunnyvale, CA, USA). High mass
resolution was used for peptide identification. The reverse
phase-LC system consisted of a peptide Cap-Trap cartridge (Acclaim
C18 Pepmap100; 500 µm х 5 mm; grain size 5 µm; Dionex) and the
Acclaim C18 PepMap100 column (75 µm in diameter and 15 cm length;
grain size, 3 µm; Dionex). The sample (20 µl) was loaded onto the
trap cartridge for 1 min in 99% H2O, 1% acetonitrile
(ACN) and 0.1% formic acid (FA) and then washed for 4 min with
0.05% solution of trifluoroacetic acid (TFA) in water. The sample
was then equilibrated for 1 min with 99% H2O, 1% ACN and
0.1% FA at a flow rate of 20 µl/min for concentrating and
desalting. Peptides were subsequently eluted over 170 min from the
analytical column via the trap cartridge using a linear gradient of
100% mobile phase A (98% H2O, 2% ACN and 0.1% FA) to
100% mobile phase B (20% H2O, 80% ACN and 0.08% FA) at a
flow-rate of 0.3 µl/min using the following gradient: 0% phase B
for 6 min; 0–50% phase B for 114 min; 50–100% phase B for 30 min;
hold at 100% phase B for 15 min; 100–0% phase B in 5 min; hold at
0% phase B for 15 min.
The LTQ Orbitrap XL equipped with a NSI was used for
the MS/MS experiment in the positive ion mode, and was operated in
a data-dependent mode using Xcalibur 2.02 software (Thermo Fisher
Scientific, Inc.). The mass spectrometer settings were as follows:
Spray voltage, 1.7 kV; capillary voltage, 43 V; tube-lens voltage,
165 V; ion transfer capillary temperature, 200°C; 1 microscan for
MS1 scans at 30,000 resolution [full width at half maximum (fwhm)
at 400 m/z]; microscans for MS2 at 7500 resolution (fwhm at 400
m/z); full MS mass range, 300–2000 m/z; and MS/MS mass range,
100–2000 m/z. In each duty cycle, the 12 most abundant precursor
ions with a ≥2 charge state, were sequentially isolated to a target
value of 10,000 ions for fragmentation, using collision-induced
dissociation in a linear trap at 35% normalized collision energy
(enhanced scanning mode). The acquired ions were dynamically
excluded for 60 sec. The automatic gain control for full MS and
MS/MS was set to 1×106 and 5×104 ions,
respectively. The maximum ion accumulation time was set to 700 ms
for MS and 150 ms for MS/MS scans. Triplicate repeats were
performed for each sample.
Data analysis
The Proteome Discoverer™ software version
1.0 (Thermo Fisher Scientific, Inc.) was used to identify proteins
from mass spectrometry analysis. Proteins were searched for using
the Mascot Server software version 2.3.02 (Matrix Science, Ltd.,
London, UK). Identified proteins were sorted the Mascot Server
software according to the Multidimensional Protein Identification
Technology score and revealed peptides where P<0.05. The lists
of the identified proteins together with the LC-MS data were
uploaded to the Skyline software program (version 1.2.0.3303;
skyline.gs.washington.edu/labkey/project/home/software/Skyline/begin.view)
to obtain the peak area of peptides in each sample. The sum of the
peak areas of all identified peptides in each sample was calculated
and normalized to the overall area of all identified proteins in
the sample.
Chemicals and reagents
HPLC gradient grade ACN was purchased from BDH
Prolabo (VWR Chemicals, Radnor, PA, USA) and FA was purchased from
Merck Millipore. Urea (ultra-pure for molecular biology), ammonium
bicarbonate (ultra-pure), dithiothreitol (ultra-pure for molecular
biology), IAA, TFE (Reagent Plus), acetic acid (HAc), protease
inhibitor cocktail, mammalian cell lysis kit, phosphate buffered
saline (PBS) and trypsin (modified, proteomics grade) were obtained
from Sigma-Aldrich (Merck Millipore).
NH4HCO3, (ultra-pure), TCEP and TFA (for
protein sequence analysis) were purchased from Fluka
(Sigma-Aldrich; Merck Millipore). HCl (purum) and KCl (purum) were
obtained from Chimmed, Inc. (Moscow, Russia). Water used in this
research was prepared using the Milli-Q system (Merck
Millipore).
Animals
The migration activity of HSCs in animals with
glioblastoma was evaluated. A total of 50 adult female Wistar rats
(age, three months; weight, <220 g) from the vivarium of the AV
Zhirmunsky Marine Biology Institute (Vladivostok, Russia) were used
for the purposes of this study. The animals were kept and cared for
in accordance with the standards of good laboratory practice. The
animals were maintained at room temperature (20°C; relative
humidity, 60%), with natural day and night cycles and with access
to water and food ad libitum. The present study was approved
by the Ethics Committee at the A.V. Zhirmunsky Institute of Marine
Biology. The control group without treatment included 25 animals
with implantation of glioma C6 cells into the brain. Rats in the
treatment group (n=25) underwent a stereotactic implantation of C6
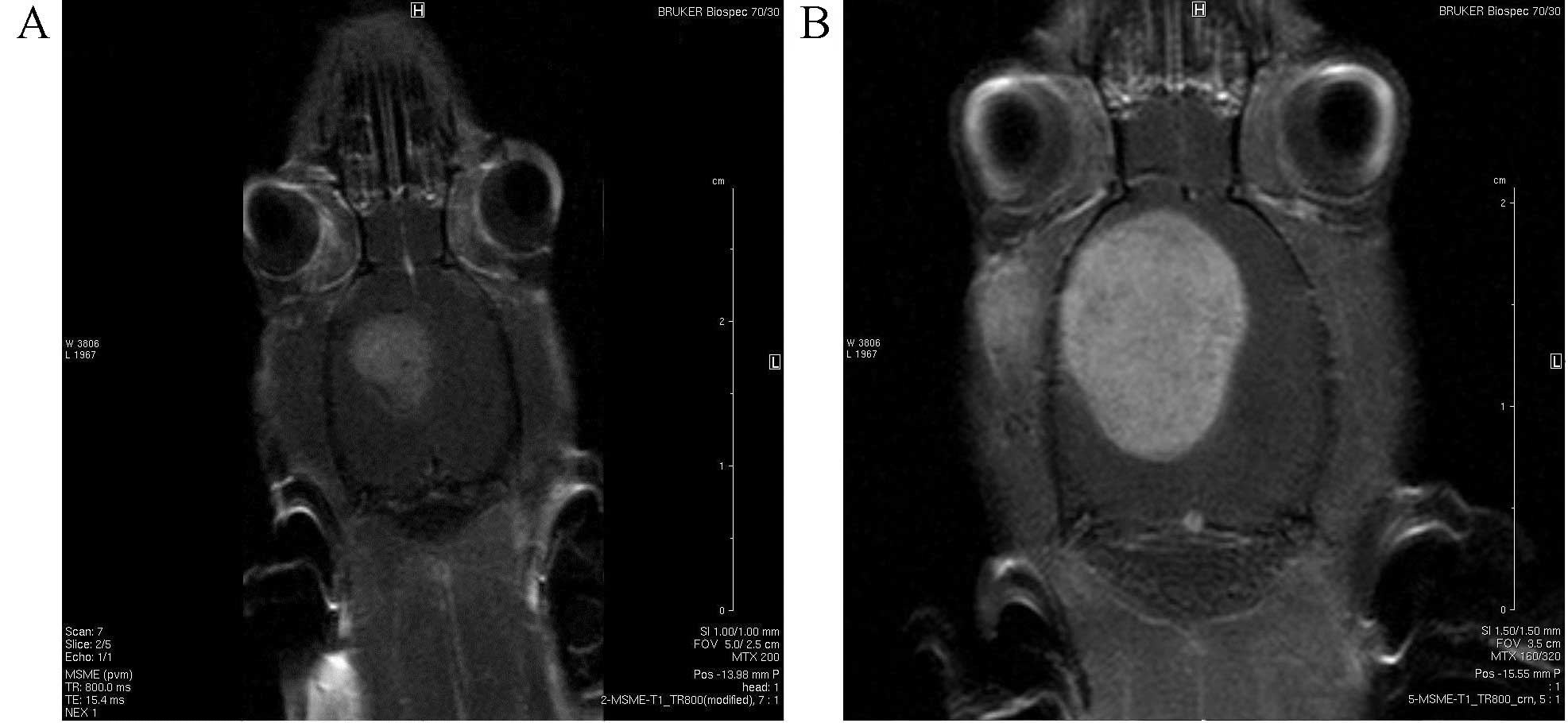
glioblastoma cells into the brain. Rats were examined by magnetic
resonance imaging (MRI; Bruker BioSpec Pharma Scan 7T; Bruker
Corporation, Billerica, MA, USA) at different stages to determine
the presence of tumors, before 5 rats from each group were subject
to histological analysis on days 7, 14, 21 and 28.
Implantation of rat glioma cells
C6 rat glioma cells were cultured as described
above, and analyzed by immunofluorescence. Cells were fixed in 4%
paraformaldehyde (PFA), washed in 0.1М PBS (pH 7.4) with 0.2% Tween
20 and 0.2% TritonХ-100 (Sigma-Aldrich; Merck Millipore) and
nonspecific binding was blocked with 0.3% bovine serum albumin
(cat. no. B4287-25G; Sigma-Aldrich; Merck Millipore). Cells were
incubated with the following primary antibodies for 24 h at 4°C:
Mouse anti-p53 (1:100; cat. no. AHO0152; Novex; Thermo Fisher
Scientific, Inc.), rabbit anti-nestin (1:100; cat. no. 5413;
Sigma-Aldrich; Merck Millipore), rabbit anti-GFAP (1:50; cat. no.
ab7260; Abcam), mouse anti-tubulin-βIII (1:100; cat. no. 7751;
Abcam), rabbit anti-S100 (1:100; cat. no. ab868; Abcam) and rabbit
anti-C-X-C chemokine receptor 4 (CXCR4; 1:100; cat. no. ab2074;
Abcam). Following washing with PBS (cat. no. 10010015; Gibco;
Thermo Fisher Scientific), Alexa488-conjugated anti-mouse (1:500;
cat. no. A11059; Novex; Thermo Fisher Scientific, Inc.) and
Alexa633-conjugated anti-rabbit (1:500; cat. no. A21071; Novex;
Thermo Fisher Scientific, Inc.) secondary antibodies were applied
for 2 h at 37°C. The nuclei were stained with
4′,6-diamidino-2-phenylindole (cat. no. D1306; Molecular Probes;
Thermo Fisher Scientific, Inc.) and following washing mounted in
Мowiol (cat. no. 324590; Sigma-Aldrich; Merck Millipore). Control
samples were stained without using primary antibodies and were
visualized under a fluorescence microscope.
Rats underwent stereotactic intracranial
implantation of C6 rat glioma cells (Narishige, Tokyo, Japan). Rats
were anesthetized with an intraperitoneal injection of Zoletil 100
(Virbac, Carros, France) and Rometar (Bioveta, Ivanovice na Hané,
Czech Republic) at a 1:4 ratio for 10 mg/kg body mass. Soft tissues
were dissected under sterile conditions, and a burr hole was made
with a microdrill (Harvard Bioscience, Inc., Holliston, MA, USA).
C6 glioblastoma cells (0.2×106) were injected with a
Hamilton syringe into caudoputamen area based on the coordinates
from the rat brain atlas (18):
Anterior-posterior, −1; medial-lateral, 3.0; dorsal-ventral, 4.5;
to a depth of 5 mm and a rate of 3 µl/min. Tumor development was
monitored using MRI.
Migration of HSCs in vivo
HSCs were first washed with PBS, then twice with
DMEM containing 10% FBS and 1% penicillin/streptomycin.
Subsequently, cells were centrifuged at 800 × g for 10 min
at 20°C. The suspension was put through a 10-µm nylon filter. Cells
were assessed by flow cytometry, following staining with the
tracker dyes Vybrant®
carboxyfluoresceindiacetatesuccinimidyl ester (CFDA-SE; Molecular
Probes; Thermo Fisher Scientific, Inc.; excitation, 488 nm;
emission, 492⁄517 nm) and Cell Tracker™ Red CMTPX (excitation, 546
nm; emission, 577/602 nm). The central tail vein of rats was
catheterized for 1 min, and all animals were injected with labeled
HSCs.
To conduct a histological analysis, 7 or 14 days
following transplantation of glioma C6 cells into rat brains, the
rats were deeply anesthetized and perfused transaortically by
introducing a solution of 4% PFA. After fixing, the rat skull was
opened, the brain was removed and fixed in 4% PFA. Serial brain
sections (20- and 40-µm thick) were prepared on a cryostat, and
7-µm thick paraffin sections were prepared on a microtome. The
sections were stained with hematoxylin and eosin. The sections were
observed under a Axio Scope A1 light microscope (Zeiss GmbH, Jena,
Germany) or a Zeiss LSM 710 META confocal microscope (Zeiss GmbH),
and images were captured using an AxioCam ICc3 digital camera
(Zeiss GmbH). Cell counting was performed using ImageJ software
(version, 1.47; National Institutes of Health, Bethesda, MD,
USA).
Statistical analysis
Statistical analysis of data was performed using the
GraphPad Prism data analysis software (version 5.01; GraphPad
Software, Inc., La Jolla, CA, USA). Data are presented as the mean
± standard deviation. The Student's t-test and Mann Whitney
U tests were used to compare groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
NSCs demonstrated the greatest ability
to migrate in vitro and more CSCs results in increased
migration
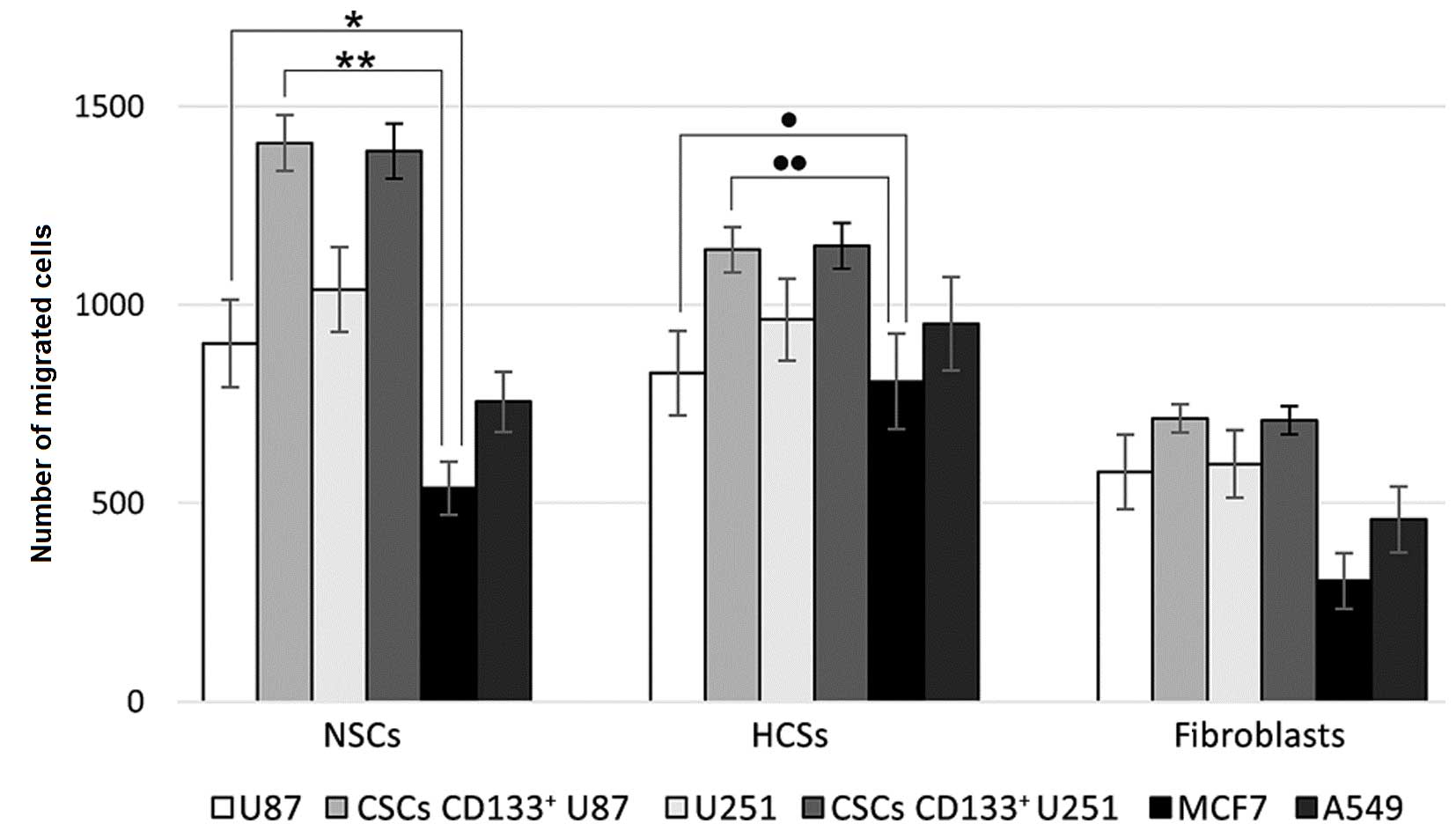
The results of comparative analysis of cell
migration activity in mixed cultures are presented in Fig. 2. NSCs migrated to a greater extent
than HSCs and fibroblasts, dependent on the type of tumor cells
present in the mixed culture. NSCs migrated more actively to the
CSCs of two glioblastoma types. Non-separated glioblastoma cells
had reduced NSCs migration-inducing ability, and tumor cells of
non-neural origin were even less able to induce migration of
NSCs.
HSCs had a slightly reduced migration activity to
glioblastoma cells compared with NSCs; however, they demonstrated
an increased migration rate towards tumor cells of ectodermic
origin. Differentiated fibroblasts migrated towards tumor cells;
however, at a reduced rate compared with NSCs and HSCs. In general,
CD133+ CSCs derived from U87 and U251 cell lines induced
migration to the greatest extent.
It is currently thought that CSCs are responsible
for the key properties of glioblastoma, namely treatment resistance
and invasive potential. In the present study, the proportion of
CSCs in glioblastoma cultures was low. The population of
CD133+ cells was 1.3±2.8% of total U87 cells and
3.4±2.6% of total U251 human glioblastoma cells. The direct
correlation between the number of CSCs in mixed cultures and the
ability to attract stem cells may indicate that that the processes
of initiation and maintaining the migration towards the tumor are
supported by the subpopulation of stem cells within the tumor.
Notably, the present study revealed a relatively
weak ability of mammary cancer and lung cancer cell cultures to
attract SCs. Mammary cancer is a hormone-dependent malignant tumor
that is surpassed in metastasis and lethality only by lung
cancer, the latter being a leading cause of cancer mortality. The
decreased ability to attract SCs may be associated with multiple
passages of this line in vitro, resulting in the loss of a
number of important signaling mechanisms.
Notably, NSCs had the greatest ability to migrate
toward tumors of neuroepithelial origin, whereas their migration to
tumors of other tissues was reduced compared with HSCs. Migration
activity may therefore be greater between cells with a common
histogenetic source and, thus, similar transcription and proteome
profiles.
HSCs indicate lower numbers of
identical proteins, however, show improved migration to
CD133+ cells
Following cell lysis, the quantity of common
proteins in lysates was as follows: NSCs (sample 1), 2032±85 µg/ml;
U87 line CSCs (sample 2), 2150±360 µg/ml; and HSCs (sample 3),
3198±281 µg/ml. The processing of LC-MS data by the Mascot Server
program identified 1,664 proteins in all samples. Subsequent to
processing with Skyline the following levels were detected: Sample
11,447 proteins based on 11,176 peptides (protein molecular weight,
3.53–3,908.10 kDa); sample 2, 1,225 proteins based on 13,674
peptides (protein molecular weight, 4.60–3,908.10 kDa); and sample
3, 842 proteins based on 10,932 peptides (protein molecular weight
5.02–1,017.07 kDa). The dynamic range for identified proteins is 7
orders, which allowed the identification of low-copy-number
proteins.
Proteins identified in only one cell type were
eliminated from proteome profiles of the compared groups. Thus,
1,659 proteins were divided into comparison group A for NSCs and
CSCs (1,052 proteins) and comparison group B for HSCs and CSCs (607
proteins). The results in comparison group A (63.4% identical
proteins) revealed the similarity of the morphofunctional
phenotypes of NSCs and CSCs in human glioblastoma; these two cells
represent a certain cell type with a specific differentiation and
localization in the central nervous system. Comparison group B had
only 607 (36.6%) identical proteins in analyzed proteomes.
The results suggest a close association between
glioblastoma CSCs and NSCs, providing an explanation for the
increased migration; however, this does not render them a potential
treatment tool. The significant differences between CSCs and HSCs
proteomes indicate that HSCs are involved in the neoplastic process
in the human brain to a much lesser extent; therefore, they have
retained their regulation properties. The ability of HSCs to
migrate towards CD133+ U87 and U251 cells means they are
a potential treatment tool for glioblastoma CSCs treatment.
Therefore, the evaluation of migration potential in vivo was
conducted with HSCs only.
HSCs migrate to glioma cells in
vivo
Immunofluorescence of C6 glioblastoma cells revealed
that 98.8±13.3% of cells expressed nestin and 82.3±16.4% expressed
the mutant p53 protein. A proportion of cells stained positive for
CXCR4 (53.7%), S100 calcium binding protein (9.4%), GFAP (7.2%) and
βIII-tubulin (9.3%). Stereotactic implantation of glioma cells into
a rat brain led to rapid tumor development visualized through MRI
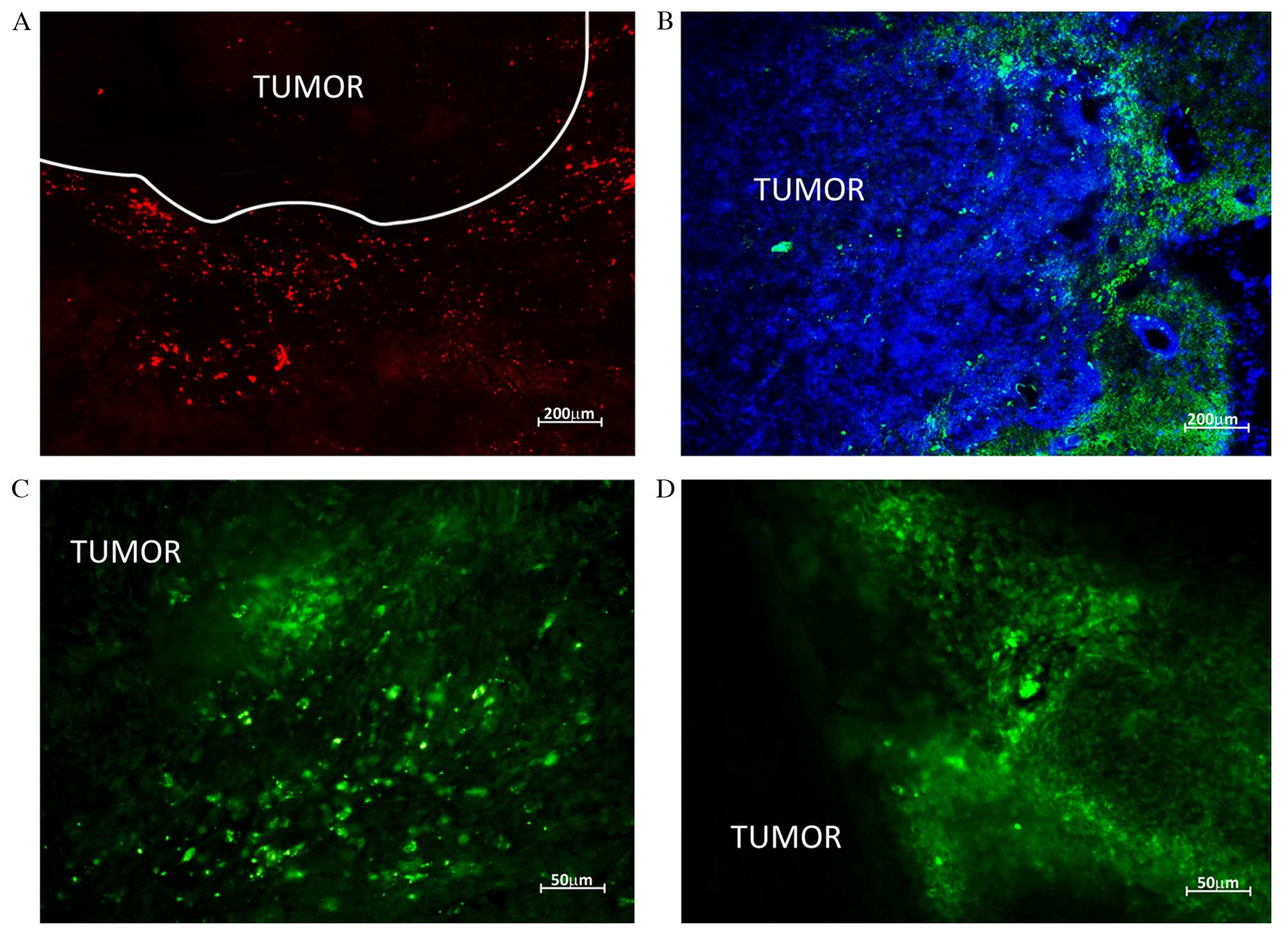
(Fig. 3).
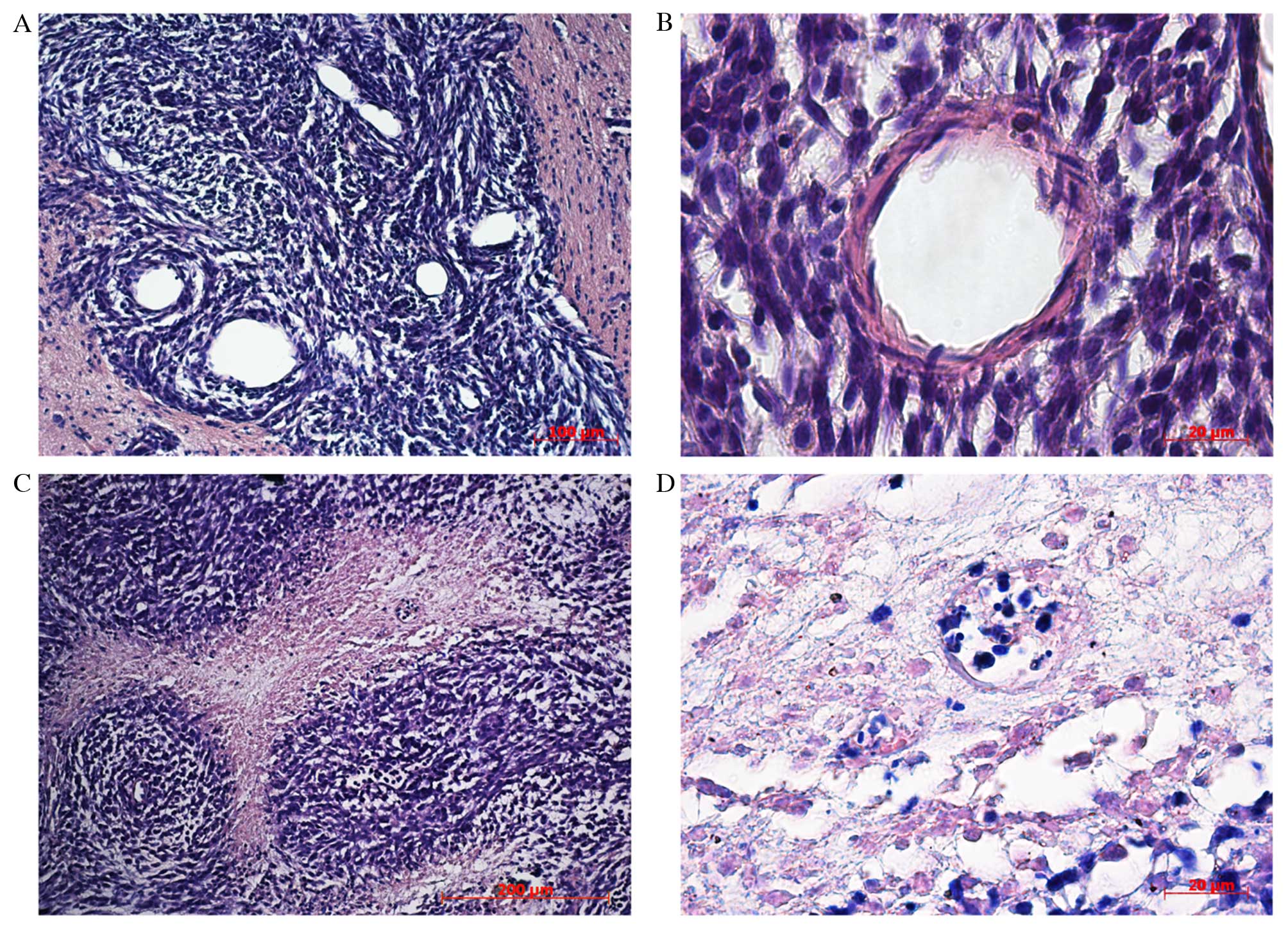
Histological analysis of the tumor revealed cells of
different shapes with varying numbers of nuclei. The tumor tissue
contained multiple microvessels, suggesting high-speed metabolism.
Along the tumor periphery glioma cells invaded the distrophically
altered brain parenchyma. The cells were clustered at a distance
from the primary node creating conglomerates that became secondary
satellite tumors with a central blood vessel. On observation day
20, tumor cells in neoplastic tissue began to die. Necrosis in the
center of the tumor node appeared, at a distance from the feeding
blood vessels, demonstrating the inability of the blood supply
system to satisfy the requirements of fast growing cells. The tumor
cells formed thick node-like clusters around the feeding blood
vessel against a background of necrosis (Fig. 4).
Injecting the animals with fluorochrome-labelled
HSCs allowed their migration to be tracked in the brain and
parenchymal organs. The greatest number of HSCs during the whole
observation term was recorded in the brain. At 7 days following
HSCs injection fluorescent areas were observed in brain parenchyma
in close proximity to the tumor; the size of fluorescent objects
was equal to the size of transplanted cells (10–20 µm). Upon
reaching the neoplastic node, HSCs clustered along the border with
normal tissue (>100 cells visible) and by means of diffusion
penetrated the tumor tissue. HSCs were clearly visible along the
tumor borders and in the center of the neoplasm 14 days following
injection (Fig. 5).
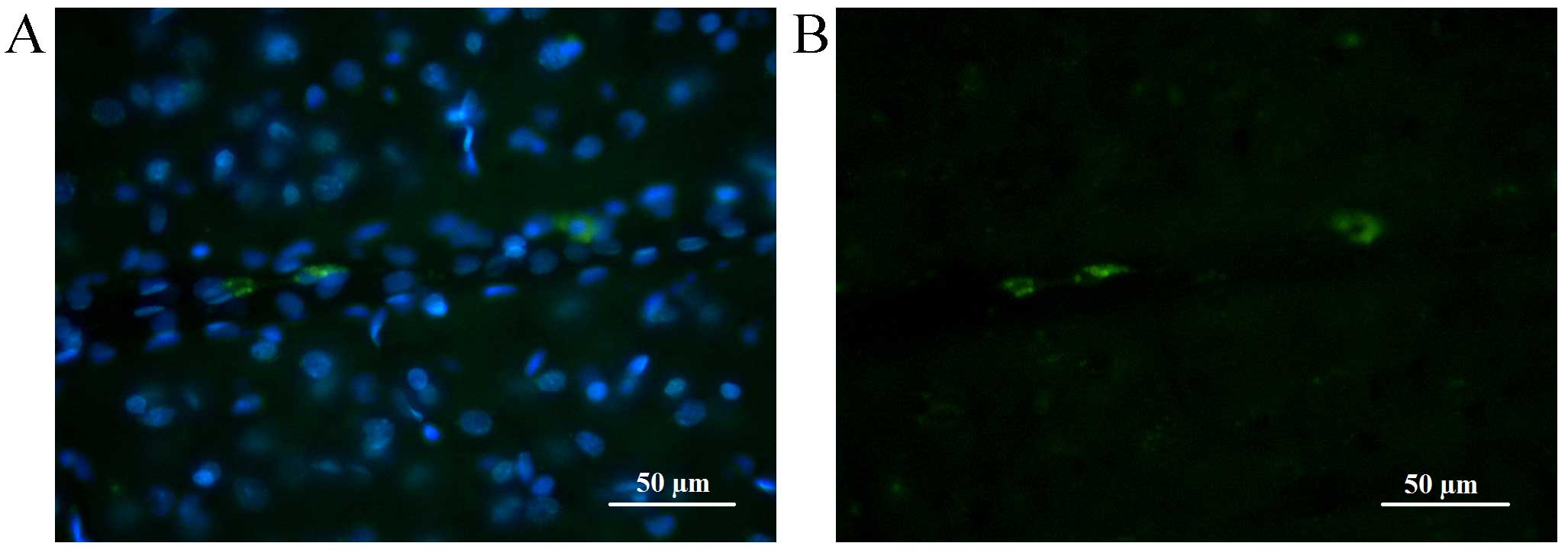
Fluorescence of CFDA-SE stained cells lasted longer
than that of CMTPX. At seven days following injection, in the brain
of the control group animals (without injected tumor), the majority
of transplanted cells were located close to or inside the
parenchymal vessel walls or in brain parenchyma where they
exhibited a long-lasting fluorescence with spectrum characteristics
distinct from the autofluorescence of brain tissue (Fig. 6).
Parenchymal organs of experimental group animals
revealed the singular fluorescent objects of the required size in
lung, spleen and bone marrow tissues 7 days following injection. On
day 14 there were no signs of transplanted cells in rats of control
and experimental groups. The majority of analyzed organs had
autofluorescent cells, the intensity of which was evaluated based
on the same parameters for the control and the experimental
group.
Lung, spleen and liver tissues revealed weak
fluorescence; however, the spectrum was distinct from the
fluorescence of the injected cells. In kidneys this signal was
discovered in tubule epithelial cells, while in lungs it was
present in a small group of alveolocytes and cells of interalveolar
septum with an oblong or rounded shape. If an animal had
inflammation of the lung parenchyma, the number of autofluorescent
cells in that area was increased, in control and experimental group
animals (data not shown).
Discussion
Transplantation of stem cells is a primary clinical
method of treating malignant tumors due to the strong ability of
transplanted cells to migrate to the damaged area. Currently it is
the sole method that may radically improve patient prognosis in the
case of hemoblastosis. A crucial stage in treatment of
oncohematologic diseases and certain other cancer types is
radiation and high-dosage chemotherapy. By eliminating
hematopoietic cells these strategies destroy their local
microsurroundings in bone marrow, while the transplanted stem cells
will find their way to the damaged area, populate and remodel it by
creating new conditions of local microenvironment (19,20).
Targeted migration of SCs to the damaged area has
been considered only in terms of its reparative function, until
recently. This process is induced by numerous factors released in
the damaged area binding to the corresponding stem cell receptors.
The role of stromal cell-derived factor interacting with CXCR4 on
SCs is defined as is the role of stem cell factor, hepatocyte
growth factor, vascular endothelial growth factor (VEGF), monocyte
chemotactic protein, proteins inhibiting microphages migration and
>80 other factors produced by the damaged tissues (21,22).
However, stem cell migration to the neoplastic nidus
is not only a reparative phenomenon. The results of the present
study demonstrate that tumor cells are a primary source of signals
activating the targeted migration of stem cells. The ability of
neuroepitheial tumors to produce tenastin, fibronectin, laminin and
collagen of various types has been demonstrated; the production of
these proteins by the damaged tissues has been associated with the
targeted migration of SCs. In addition, an important characteristic
of glioblastoma is hypoxia, which maintains cells in a
non-differentiated state for an extended period and triggers the
expression of more >100 genes inducing SCs migration (23).
In the present study, CSCs had the greatest
stability to attract SCs. Numerous cytokines are released by CSCs,
including chitinase 3-like protein 1, a disintegrin and
metalloproteinase (ADAM) 9, ADAM10, cathepsins B and L1,
osteopontin, semaphoring 7A and other inducers of targeted
migration and stem cell homing (24,25).
In the present study, 33 released proteins were identified in the
CSC proteome that may potentially influence stem cell migration (as
presented in Table I).
 | Table I.Secreted proteins present in U87
human glioblastoma cancer stem cells. |
Table I.
Secreted proteins present in U87
human glioblastoma cancer stem cells.
| NSI, % | Protein |
|---|
| 0.018017 | CD59 molecule,
complement regulatory protein |
| 0.017455 | Niemann-Pick
disease, type C2 |
| 0.230439 | Actinin, alpha
1 |
| 0.291456 | Actinin, alpha
4 |
| 0.701277 | Annexin A2
pseudogene 3; annexin A2; annexin A2 pseudogene 1 |
| 0.023723 | Calmodulin 3
(phosphorylase kinase, delta); calmodulin 2 (phosphorylase kinase,
delta); calmodulin 1 (phosphorylase kinase, delta) |
| 0.059753 | Calumenin |
| 0.009112 | Cathepsin B |
| 0.167202 | Cathepsin D |
| 0.012532 | Collagen, type VI,
alpha 3 |
| 0.006565 | Dermcidin |
| 0.084246 | Gelsolin
(amyloidosis, Finnish type) |
| 0.000635 | Granulin |
| 0.60557 | Heat shock 60 kDa
protein 1 (chaperonin) pseudogene 5; heat shock 60 kDa protein 1
(chaperonin) pseudogene 6; heat shock 60 kDa protein 1 (chaperonin)
pseudogene 1; heat shock 60 kDa protein 1 (chaperonin) pseudogene
4; heat shock 60 kDa protein 1 (chaperonin) |
| 0.01018 | High density
lipoprotein binding protein |
| 0.157602 | Macrophage
migration inhibitory factor (glycosylation-inhibiting factor) |
| 0.029513 | Peroxiredoxin
4 |
| 0.083528 |
Phosphatidylethanolamine binding protein
1 |
| 0.103943 | Plasminogen |
| 0.182445 | Prosaposin |
| 0.015039 | Reticulon 3 |
| 0.259942 |
Ribonuclease/angiogenin inhibitor 1 |
| 0.007125 | Serpinpeptidase
inhibitor, clade E (nexin, plasminogen activator inhibitor type 1),
member 2 |
| 0.084381 | Superoxide
dismutase 1, soluble |
| 0.054239 | Tyrosyl-tRNA
synthetase |
| 0.004402 | Zinc finger protein
91 homolog (mouse); ZFP91-CNTF readthrough transcript; ciliary
neurotrophic factor |
Notably, NSCs were markedly more active in relation
to glioblastoma CSCs. During tumor development in the nervous
tissue NSCs and CSCs develop complex interactions. Glioblastoma
development inhibits NSC proliferation in germinal brain areas in
older experimental animals (26).
However, NSCs may be a source of cells recruited by the tumor for
proliferation and metabolism. NSCs and glioblastoma CSCs have
numerous similar morphological and biochemical properties; they
actively proliferate in vitro and create neurospheres
(gliomaspheres), and they may differentiate following the
introduction of various factors, including VEGF, nerve growth
factor and bone morphogenetic protein into the medium. Nestin is
the most important marker of NSCs, and this marker was present in
the majority of C6 glioblastoma cells that were used in the present
study. NSC descendants are neural progenitor cells that are
constantly migrating from subventricular zone; this partially
accounts for their high mobility in the present study (27,28).
NSCs may transform into CSCs due to the
environmental conditions of the tumor. The high level of similarity
(63.5%) between NSCs and CSCs demonstrated in the present study
supports this hypothesis. By contrast, HSCs with a decreased
mobility to CSCs in mixed cultures have fewer proteins expressed by
CSCs, suggesting that this cell type is less prone to neoplastic
transformation in neural tumors. HSCs may therefore be a potential
tool for developing technologies aimed at controlling the activity
of glioblastoma CSCs.
The standard method for evaluating the migration of
exogenous cells in experimental animals is fluorescence laser
microscopy (29); however,
implanted SCs in animal organs do not always demonstrate definite
results. HSCs fluorescence in parenchymal organs has been described
for rats injected with Lewis, Ehrlich and Fisher adenocarcinoma
along with certain other models; however, fluorescent cells were
identified in the tumor tissue. Clusters of transplanted cells in
tumor-carrying organism are typically observed in lung, spleen and
bone marrow. This fact is frequently explained by the small
quantity of injected cells and the obstacles produced by
autofluorescence of the examined tissues, or the inadequacy of the
selected cell type and animal species for the experiment (30).
The targeted migration of SCs to the neoplastic
tumor has been demonstrated using experimental glioblastoma models
for hematopoietic, neural, fetal and embryonic SCs (31,32).
The ability to attract SCs may be a key glioblastoma property. This
may be associated with the high concentration of CSCs in the
population of cancer cells.
Interaction of normal and cancer SCs in the
neoplasia area creates certain conditions resulting in suppression
of tumor proliferation, angiogenesis and metastasis, stimulation of
inflammation and initiation of apoptosis. Certain underlying
mechanisms of this interaction have been described, including
intensification of specific cytokines production, tumor cell cycle
arrest in G1 phase, proapoptotic modification of SCs
increasing the efficiency of cytoplasmic protein exchange and the
by-stander effect and molecular adhesion phenomenon (33–35).
The phenomenon of molecular adhesion was first
described by Aboody et al (36) in 2000. It is based on the SCs
unique ability to follow the tumor cell into the brain parenchyma
using its cytokine trail, reach and adhere to it. Our laboratory
has described the mechanism underlying intercellular communication
between stem and tumor cells via exosomes, enabling active
cytoplasmic protein exchange that suppresses tumor cell
proliferation (37). It can be
assumed that modifying the proteome profile of HSCs may facilitate
the generation of cell systems that can trigger targeted apoptosis
in neoplastic stem cells.
In conclusion, the results of the present study
demonstrate that CSCs demonstrate the greatest ability to attract
normal SCs, out of all tumor cells assessed. The differences in
cell proteomes suggest HSCs as a potential tool for interacting
with glioblastoma CSCs. Following the injection of experimental
animals with glioblastoma, HSCs migrated to the brain hemisphere
containing the tumor and penetrated the neoplastic tissue. Further
research regarding the application of hematopoietic stem cells in
the treatment of malignant tumors of the central nervous system is
required, however, the results of the present study form the basis
for the production of novel antitumor therapies.
Acknowledgements
The present study was conducted with financial
support from the Russian Science Foundation (project no.
14-15-00084).
References
|
1
|
Omuro A and DeAngelis LM: Glioblastoma and
other malignant gliomas: A clinical review. JAMA. 310:1842–1850.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stupp R and Hegi ME: Brain cancer in 2012:
Molecular characterization leads the way. Nat Rev Clin Oncol.
10:69–70. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stupp R and Weber DC: The role of radio-
and chemotherapy in glioblastoma. Onkologie. 28:315–317.
2005.PubMed/NCBI
|
|
4
|
Soltanian S and Matin MM: Cancer stem
cells and cancer therapy. Tumour Biol. 32:425–440. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tabatabai G and Weller M: Glioblastoma
stem cells. Cell Tissue Res. 343:459–465. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Taal W, Bromberg JE and van den Bent MJ:
Chemotherapy in glioma. CNS Oncol. 4:179–192. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lathia JD, Mack SC, Mulkearns-Hubert EE,
Valentim CL and Rich JN: Cancer stem cells in glioblastoma. Genes
Dev. 29:1203–1217. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zaridze DG: Carcinogenesis. Medicine;
Zaridze D.G.: pp. 1–567. 2004, PubMed/NCBI
|
|
9
|
Hide T, Makino K, Nakamura H, Yano S, Anai
S, Takezaki T, Kuroda J, Shinojima N, Ueda Y and Kuratsu J: New
treatment strategies to eradicate cancer stem cells and niches in
glioblastoma. Neurol Med Chir (Tokyo). 53:764–772. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Duesberg P, Mandrioli D, McCormack A and
Nicholson JM: Is carcinogenesis a form of speciation? Cell Cycle.
10:2100–2014. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pecchia I, Dini V, Ricci-Vitiani L,
Biffoni M, Balduzzi M, Fratini E, Belli M, Campa A, Esposito G,
Cirrone G, et al: Glioblastoma stem cells: Radiobiological response
to ionising radiations of different qualities. Radiat Prot
Dosimetry. 166:374–378. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bryukhovetskiy IS, Mischenko PV, Tolok EV,
Zaitcev SV, Khotimchenko YS and Bryukhovetskiy AS: Directional
migration of adult hematopoietic progenitors to C6 glioma in vitro.
Oncol Lett. 9:1839–1844. 2015.PubMed/NCBI
|
|
13
|
Bryukhovetskiy I, Bryukhovetsky A,
Khotimchenko Y, Mischenko P, Tolok E and Khotimchenko R:
Combination of the multipotent mesenchymal stromal cell
transplantation with administration of temozolomide increases
survival of rats with experimental glioblastoma. Mol Med Rep.
12:2828–2834. 2015.PubMed/NCBI
|
|
14
|
Studeny M, Marini FC, Dembinski JL,
Zompetta C, Cabreira-Hansen M, Bekele BN, Champlin RE and Andreeff
M: Mesenchymal stem cells: Potential precursors for tumour stroma
and targeted-delivery vehicles for anticancer agents. J Natl Cancer
Inst. 96:1593–1603. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mooney R, Roma L, Zhao D, Van Haute D,
Garcia E, Kim SU, Annala AJ, Aboody KS and Berlin JM: Neural stem
cell-mediated intratumoural delivery of gold nanorods improves
photothermal therapy. ACS Nano. 8:12450–12460. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kanojia D, Balyasnikova IV, Morshed RA,
Frank RT, Yu D, Zhang L, Spencer DA, Kim JW, Han Y, Yu D, et al:
Neural stem cells secreting anti-HER2 antibody improve survival in
a preclinical model of HER2 overexpressing breast cancer brain
metastases. Stem Cells. 33:2985–2994. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pittenger MF: Mesenchymal stem cells from
adult bone marrow. Methods Mol Biol. 449:27–44. 2008.PubMed/NCBI
|
|
18
|
Paxinos G and Watson C: The Rat Brain in
Stereotaxic Coordinates. 6th. Academic Press; Cambridge, MA: pp.
1–456. 2007
|
|
19
|
Shipounova IN, Petinati NA, Bigildeev AE,
Zezina EA, Drize NI, Kuzmina LA, Parovichnikova EN and Savchenko
VG: Analysis of results of acute graft-versus-host disease
prophylaxis with donor multipotent mesenchymal stromal cells in
patients with hemoblastoses after allogeneic bone marrow
transplantation. Biochemistry (Mosc). 79:1363–1370. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Amson R, Karp JE and Telerman A: Lessons
from tumour reversion for cancer treatment. Curr Opin Oncol.
25:59–65. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Naaldijk Y, Johnson AA, Ishaka S, Meiseld
HJ, Hohausd C and Stolzingb A: Migrational changes of mesenchymal
stem cells in response to cytokines, growth factors, hypoxia and
aging. Exp Cell Res. 338:97–104. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bryukhovetskiy IS, Bryukhovetskiy AS,
Mischenko PV and Khotimchenko YS: Role of systemic migration
mechanisms and stem cell homing in the development of malignant
tumours of the central nervous system and creating new methods of
antitumour therapy. Russian Biotherapeutic Journal. 4:3–12.
2013.(In Russian).
|
|
23
|
Herrera-Perez M, Voytik-Harbin SL and
Rickus JL: Extracellular matrix properties regulate the migratory
response of glioblastoma stem cells in three-dimensional culture.
Tissue Eng Part A. 21:2572–2582. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sangar V, Funk CC, Kusebauch U, Campbell
DS, Moritz RL and Price ND: Quantitative proteomic analysis reveals
effects of epidermal growth factor receptor (EGFR) on
invasion-promoting proteins secreted by glioblastoma cells. Mol
Cell Proteomics. 13:2618–2631. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Formolo CA, Williams R, Gordish-Dressman
H, MacDonald TJ, Lee NH and Hathout Y: Secretome signature of
invasive glioblastoma multiforme. J Proteome Res. 10:3149–3159.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Walzlein JH, Synowitz M, Engels B,
Markovic DS, Gabrusiewicz K, Nikolaev E, Yoshikawa K, Kaminska B,
Kempermann G, Uckert W, et al: The antitumourigenic response of
neural precursors depends on subventricular proliferation and age.
Stem Cells. 26:2945–2954. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Miska J and Lesniak MS: Neural stem cell
carriers for the treatment of glioblastoma multiforme.
EBioMedicine. 2:774–775. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Neradil J and Veselska R: Nestin as a
marker of cancer stem cells. Cancer Sci. 106:803–811. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Furia L, Pelicci P and Faretta M: Confocal
microscopy for high-resolution and high-content analysis of the
cell cycle. Curr Protoc Cytom. 70:7.42.1–7.42.14. 2014. View Article : Google Scholar
|
|
30
|
Motaln H and Turnsek TL: Cytokines play a
key role in communication between mesenchymal stem cells and brain
cancer cells. Protein Pept Lett. 22:322–331. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kidd S, Spaeth E, Dembinski JL, Dietrich
M, Watson K, Klopp A, Battula VL, Weil M, Andreeff M and Marini FC:
Direct evidence of mesenchymal stem cell tropism for tumour and
wounding microenvironments using in vivo bioluminescent imaging.
Stem Cells. 27:2614–2623. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Moore XL, Lu J, Sun L, Zhu CJ, Tan P and
Wong MC: Endothelial progenitor cells' ‘homing’ specificity to
brain tumours. Gene Ther. 11:811–818. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Aboody KS, Najbauer J, Metz MZ, D'Apuzzo
M, Gutova M, Annala AJ, Synold TW, Couture LA, Blanchard S, Moats
RA, et al: Neural stem cell-mediated enzyme/prodrug therapy for
glioma: Preclinical studies. Sci Transl Med. 5:184ra592013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu Y, Kobayashi A, Maeda T, Fu Q, Oikawa
M, Yang G, Konishi T, Uchihori Y, Hei TK and Wang Y: Target
irradiation induced bystander effects between stem-like and
non-stem-like cancer cells. Mutat Res. 773:43–47. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chan JK and Lam PY: Human mesenchymal stem
cells and their paracrine factors for the treatment of brain
tumours. Cancer Gene Ther. 20:539–543. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Aboody KS, Brown A, Rainov NG, Bower KA,
Liu S, Yang W, Small JE, Herrlinger U, Ourednik V, Black PM, et al:
Neural stem cells display extensive tropism for pathology in adult
brain: Evidence from intracranial gliomas. Proc Natl Acad Sci USA.
97:12846–12851. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bryukhovetskiy IS, Mischenko PV, Tolok EV,
Khotimchenko YS, Zaitcev SV and Bryukhovetskiy AS: Hematopoietic
stem cells with induced apoptosis effectively inhibit glioma cell
growth in vitro, but started new mechanism of tumour stem cells.
Genes and Cells. 9:70–75. 2014.
|