Introduction
Bisphosphonates (BPs) are pharmacological inhibitors
of bone resorption that have been in use for >30 years and are
commonly used to treat diseases associated with excessive bone
loss, including osteoporosis, multiple myeloma and complications
associated with bone cancer metastases (1–3).
Zoledronate is a third-generation BP, approved for
the treatment of cancer-induced bone diseases, with potent
therapeutic effects in suppressing osteoclastic activity and
resorptive bone loss (4,5). However, long-term use of Zoledronate
can result in BP-associated osteonecrosis of the jaw (BP-ONJ)
(6–8). BP-ONJ is most commonly associated
with patients with malignancies that receive high-dose intravenous
BP, occurring significantly less frequently in patients with
osteoporosis that receive orally-administered BP (9). Previous studies have demonstrated
that Zoledronate can promote osteoclast apoptosis by inhibiting
enzymes of the mevalonate pathways (10). However, in addition to osteoclast
inhibition, osteoblasts and osteocytes are also inhibited by
Zoledronate, leading to suppression of bone remodeling (11,12).
Additionally, BP-ONJ has been associated with BP-induced
anti-angiogenic effects; failed wound healing is occasionally
observed following protracted Zoledronate use, which may result in
secondary necrotic bone (13,14).
Clinical and experimental studies have also indicated that
Zoledronate induces endothelial cell apoptosis and subsequently
reduces angiogenesis (13,15,16).
Notably, Zoledronate can reduce the number of endothelial cells
within alveolar bone, particularly following tooth extraction, due
to greater accumulation of Zoledronate here than in other sites of
the skeleton (11,17). However, the regulatory mechanism by
which Zoledronate promotes endothelial cell apoptosis has not been
clearly elucidated at the molecular level.
Autophagy is a highly regulated process, involved in
the degradation and recycling of proteins, intracellular pathogens
and cytoplasmic organelles (18)
and is important for cell survival, differentiation and homeostasis
(19,20). Previous studies have demonstrated
Zoledronate-induced autophagy in various tumor cells (18,21–23).
However, the ability of Zoledronate to induce autophagy in
endothelial cells remains unknown. In addition, autophagy has been
suggested to be involved in crosstalk with apoptosis by inhibiting
or promoting the process (21,24).
Thus, the present study hypothesized that Zoledronate may induce
apoptosis in human umbilical vein endothelial cells (HUVECs),
partially, by affecting autophagy. In addition to confirmation of
Zoledronate-induced apoptosis in HUVECs, the present study also
aimed to provide evidence of the critical function of autophagy in
Zoledronate-induced apoptosis.
Materials and methods
Reagents
Endothelial basal medium (EBM), penicillin,
streptomycin, fetal calf serum (FCS), human endothelial growth
factor β (β-ECGF), TRIzol reagent and Lipofectamine 2000 were
purchased from Invitrogen (Thermo Fisher Scientific, Inc., Waltham,
MA, USA). Zoledronate (full name,
2-(imidazole-1-yl)-hydroxy-ethylidene-1, 1-bisphosphonic acid,
disodium salt, 4.75 hydrate; molecular weight, 401.6) was purchased
from Novartis International AG (Basel, Switzerland). A stock
solution of Zoledronate (100 mM) was prepared in filter-sterilized
phosphate-buffered saline (PBS). Chloroquine, 3-methyladenine
(3-MA), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT) and Hoechst 33258 were obtained from Sigma-Aldrich
(Merck Millipore, Darmstadt, Germany).
Cell culture
Human umbilical cord veins were obtained from the
Affiliated Nanjing University Medical School and human umbilical
vein endothelial cells (HUVECs) were isolated as previously
described (25). The study was
approved by the Medical Research Ethics Committee of Medical School
of Nanjing University and informed content was obtained from 20
pregnant women giving birth between 2012 and 2014. In brief, cells
were harvested from umbilical cord by 0.125% trypsin, and then
cultured in EBM supplemented with 100 U/ml penicillin, 100 µg/ml
streptomycin, 20% FCS and 100 µg/ml EGF in an incubator with 5% CO2
and 95% air at 37°C.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) for microtubule-associated
proteins 1A/1B light chain 3B (LC3B) expression
Total RNA from HUVECs treated with various
concentrations of Zoledronate (25, 50, 75 and 100 µM) for 48 h was
isolated using TRIzol reagent according to the manufacturer's
protocols. Total RNA (1 µg) was reverse-transcribed using
SuperScript III First-Strand Synthesis system (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. Amplification was performed using Fast
SYBR® Green Master Mix Kit (Applied Biosystems; Thermo
Fisher Scientific, Inc.) in an Applied Biosystems 7300 Fast RT-PCR
system and LC3B expression was calculated using the
2-ΔΔCq method as previously described (26). A total of 35 cycles were conducted
consisting of denaturation at 95°C for 15 sec, primer annealing at
60°C for 1 min and primer extension at 72°C for 30 sec. The
specific primer sequences used in this study were as follows: LC3B,
forward, 5′-AGCAGCATCCAACCAAAATC-3′, reverse,
5′-CTGTGTCCGTTCACCAACAG-3′; 18S, forward,
5′-CGGCTACCACATCCAAGGAA-3′, reverse, 5′-CTGGAATTACCGCGGCT-3′.
Western blot analysis
Proteins from HUVECs (2×105 cells) treated with
various concentrations of Zoledronate (25, 50, 75 and 100 µM) for
48 h were isolated with radioimmunoprecipitation assay buffer
(Beyotime Institute of Biotechnology, Jiangsu, China) containing
protease and phosphatase inhibitor cocktail (Merck Millipore,
Darmstadt, Germany). The protein content of cell lysates was
determined by Bradford assay (Bio-Rad Laboratories, Inc., Hercules,
CA, USA). An equal amount of protein (50 µg) from HUVEC lysates was
separated by 10–12% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis, blotted onto Immobilon-P membranes (Merck
Millipore, Volketswil, Switzerland), blocked with 5% non-fat milk
with TBST (Tris-HCL 20 mM, NaCl 150 mM, 0.1% Tween 20) and then
incubated with antibodies against LC3B (cat. no. 3868), sequestome
1 (SQSTM1) (cat. no. 8025), Beclin-1 (cat. no. 3738), cleaved
caspase-9 (cat. no. 7237), cleaved caspase-3 (cat. no. 9579) and
β-actin (cat. no. 3700) (1:1,000; Cell Signaling Technology, Inc.,
Danvers, MA, USA) overnight at 4°C. Following incubation with the
horseradish peroxide-labeled goat anti-rabbit (cat. no. A0208) or
goat anti-mouse (cat. no. A0216) antibody (Beyotime Institute of
Biotechnology) at room temperature for 1 h, the signals were
visualized on radiographic film using enhanced chemiluminescence
reagents (Bio-Rad Laboratories, Inc.). The density of the
appropriate sized bands was quantified using ImageJ software,
version 1.41 (National Institutes of Health, Bethesda, MD,
USA).
Green fluorescent protein (GFP)-LC3
adenovirus infection and assay
Adenovirus encoding GFP-LC3 (Ad-GFP-LC3) was
obtained from Cyagen Biotechnology Co., Ltd. (Santa Clara, CA,
USA). HUVECs (2×105 cells) were cultured in EBM containing 2% FCS
and Ad-GFP-LC3 (multiplicity of infection, 100) for 3 h, following
which, the medium was replaced with fresh complete medium. HUVECs
were infected with Ad-GFP-LC3 for 24 h prior to treatment with
various concentrations of Zoledronate for a further 48 h. Following
treatment, cells were harvested and fixed in 4% paraformaldehyde
and visualized and photographed in Zeiss LSM 5 Pascal laser
scanning confocal fluorescent microscope (Zeiss AG, Oberkochen,
Germany). The average number of GFP-LC3 punctae per cell were
counted to quantify autophagy activities using ImageJ software,
version 1.41 (National Institutes of Health).
Cell viability assay
Cell viability was determined by MTT assay. HUVECs
(2×103 cells/well) were plated in 96-well culture plates and
rendered quiescent by culturing in serum-free medium overnight at
37°C. Following treatment with different concentrations of
Zoledronate for 48 h, 10 µl MTT (5 mg/ml) was added to each well,
and plates incubated for a further 4 h. The medium was removed and
100 µl of dimethyl sulfoxide was added to solubilize the formazan
crystals. Absorbance of the medium was measured at 570 nm using the
SPECTRAMax M5 reader (Molecular Devices, LLC, Sunnyvale, CA,
USA).
Cell apoptosis assay
HUVEC were treated with various concentrations of
Zoledronate (25, 50, 75 and 100 µM) for another 48 h, or pretreated
with 3-MA (5 mM) for 30 min prior to Zoledronate (100 µM)
incubation for another 48 h. Cell apoptosis was evaluated by
fluorescein isothiocyanate (FITC)-conjugated Annexin-V and
propidium iodide (PI) staining by using FITC-Annexin V and PI
double staining kit (KeyGen Biotech Co., Ltd, Nanjing, China)
according to the manufacturer's protocols. Treated or untreated
cells (2×105) were digested and suspended in 500 µl binding buffer
plus 5 µl FITC-labeled Annexin V and 5 µl PI solution. The mixtures
were incubated on ice for 10 min, and then plotted for
FITC-conjugated Annexin V and PI in a two-way dot plot to count the
apoptotic cells with flow cytometry (BD Biosciences, San Jose, CA,
USA).
Hoechst 33258 dye staining
Apoptotic morphology of HUVECs was observed by
Hoechst 33258 dye. Cells were washed twice with PBS, fixed in 4%
paraformaldehyde for 10 min, permeabilized with 0.1% Triton X-100
and incubated with 2.5 µg/µl Hoechst 33258 for 5 min at room
temperature. Stained cells were washed, and nuclei observed by
fluorescence microscopy using an IX83 inverted motorized microscope
(Olympus Corporation, Tokyo, Japan).
Small interfering RNA (siRNA)
transfection
Beclin-1 siRNA (5′-GGUCUAAGACGUCCAACAA-3′) and
non-targeting negative control siRNA (5′-UGGUUUACAUGUCGACUAA-3′;
Invitrogen; Thermo Fisher Scientific, Inc.) were transiently
transfected with Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Briefly, siRNA and Lipofectamine 2000 were mixed at ratio of 1:4,
then incubated in serum- and antibiotic-free EBM for 15 min. The
mixture was added to 70% confluence of HUVECs and swirled gently to
ensure uniform distribution. Following 3 h incubation at room
temperature, the transfection mixture was removed and fresh
complete medium was added prior to further incubation. HUVECs were
incubated for 24 h following siRNA transfection, followed by 48 h
Zoledronate (100 µM) treatment.
Statistical analysis
All data are expressed as the mean ± standard error,
and n values indicate the number of independent experiments
performed. Comparisons between multiple groups were analyzed by
one-way analysis of variance, followed by Tukey's multiple
comparison post-hoc test. All statistical analyses were performed
using SPSS software (version 17.0; SPSS Inc., Chicago, IL, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Zoledronate induces the expression of
markers for autophagy in HUVECs
qPCR analysis of HUVECs revealed a significant
dose-dependent increase in the mRNA expression levels of LC3B
following Zoledronate treatment (Fig.
1A). To further confirm autophagy in HUVECs was mediated by
Zoledronate, the conversion of LC3B-I to LC3B-II was analyzed, as
this suggests an increase in the number of autophagosomes within
cells (27). Although the protein
level of LC3B-I was marginally increased, Zoledronate treatment
significantly increased the levels of LC3B-II and the ratio of
LC3B-II/I compared with untreated cells (P<0.05; Fig. 1B). Furthermore, infection with
GFP-LC3 adenovirus revealed a dose-dependent increase in the number
of GFP puncta in the cytoplasm following Zoledronate exposure,
indicating increased transformation of LC3B-I to LC3B-II
(P<0.05; Fig. 1C and D). By
contrast, the levels of SQSTM1, which is degraded in the lysosome
following autophagosome fusion (28), were significantly decreased in
HUVECs that were treated with >50 µM Zoledronate in a
dose-dependent manner (P<0.01; Fig.
1E). Western blot analysis of another autophagy marker,
Beclin-1, which is required for the formation of autophagosomes
(29), revealed that the protein
expression level of Beclin-1 was increased dose-dependently
following exposure to Zoledronate (P<0.05; Fig. 1F). Zoledronate was, therefore,
demonstrated to induce autophagy in HUVECs.
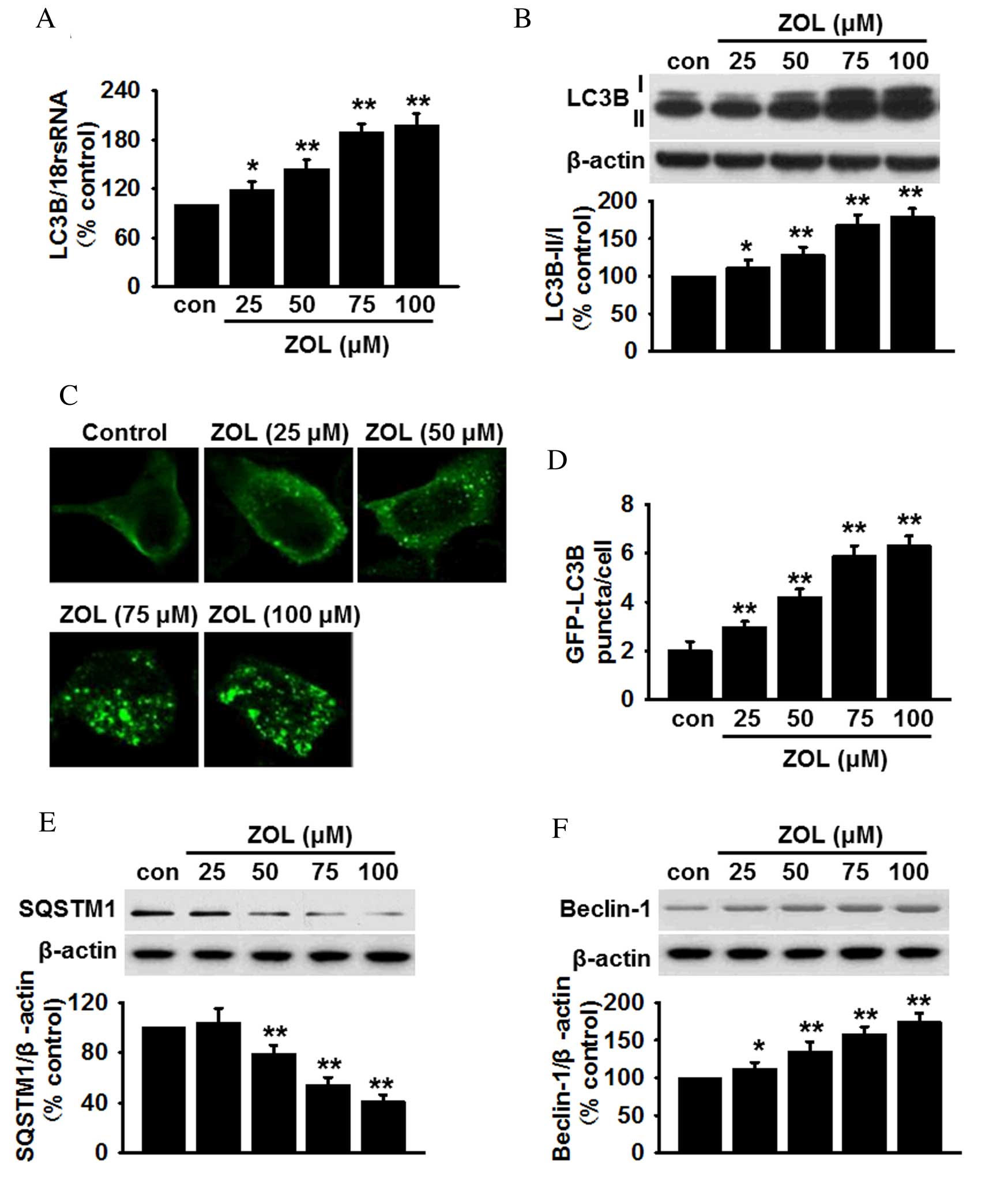 | Figure 1.ZOL induces dose dependent autophagy
in HUVECs. (A) Cells were treated with 25, 50, 75 and 100 µM ZOL
for 48 h, then the mRNA expression levels of LC3B were determined
by reverse transcription-quantitative polymerase chain reaction.
(B) The levels of LC3B-I and LC3B-II were analyzed by
semi-quantitative western blot. (C) HUVECs were infected with
Ad-GFP-LC3 adenovirus prior to ZOL treatment, following which,
GFP-LC3 punctae were observed using a laser scanning confocal
fluorescent microscope. A representative single cell exhibits LC3
punctae as a marker of autophagic vesicles. (D) Quantification of
the mean number of GFP-LC3 punctae per cell. Representative blots
and quantitative bar graphs demonstrating the expression of (E)
SQSTM1 and (F) Beclin-1, following ZOL treatment. All data are
presented as the mean ± standard error. *P<0.05, **P<0.01 vs.
untreated control, n=4–6. HUVECs, human umbilical vein endothelial
cells; LC3B, microtubule-associated proteins 1A/1B light chain 3B;
18S rRNA, 18S ribosomal RNA; Con, control; ZOL, Zoledronate;
SQSTM1, sequestome 1. |
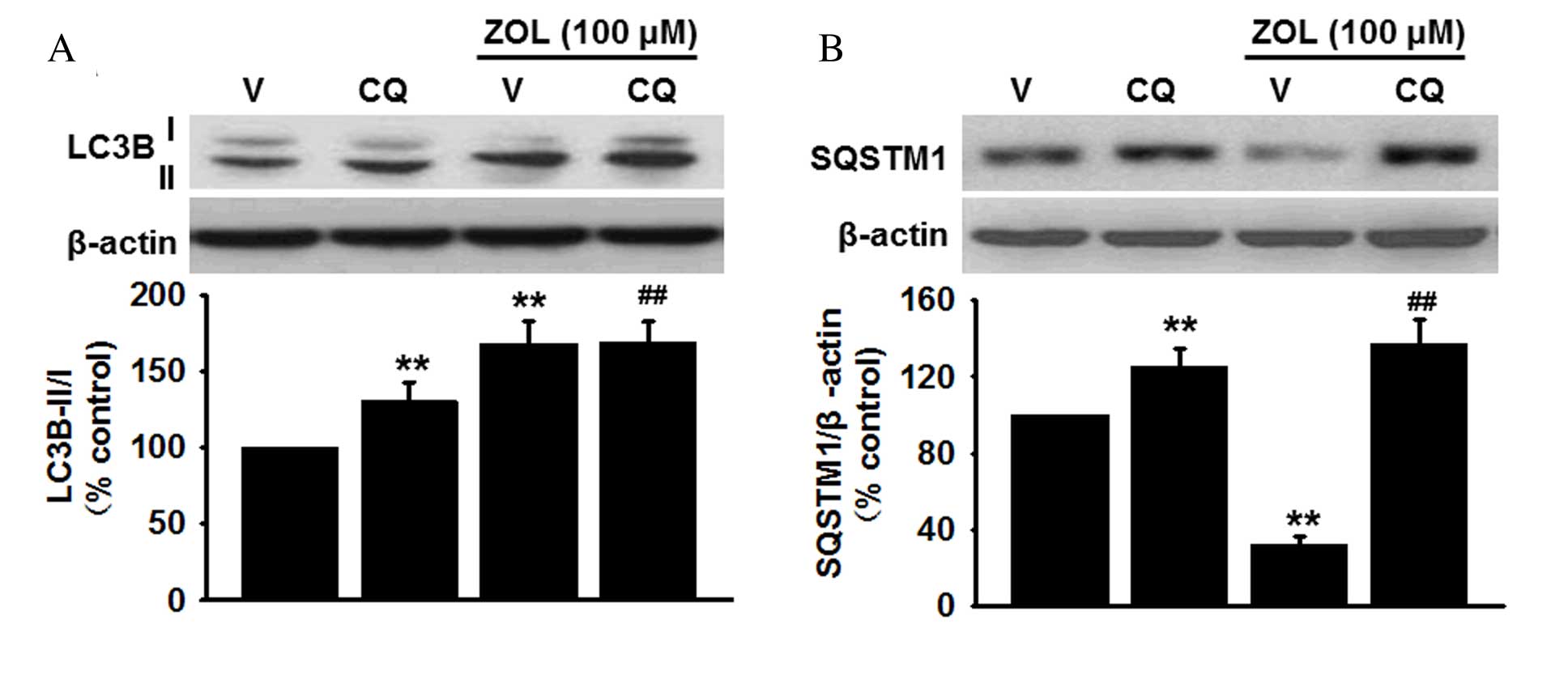
Zoledronate induces autophagic flux in
HUVECs
Increased autophagy can be attributed either to
enhanced autophagosome formation or reduction of lysosomal activity
(30). Since the increased
autophagosome formation or the decreased lysosomal degradation may
result in LC3B-II accumulation, the protein expression levels of
LC3B-II were measured in HUVECs treated with 100 µM Zoledronate in
the presence or absence of the lysosome inhibitor, chloroquine (1
µM). Western blot analysis revealed that pretreatment with
chloroquine increased basal LC3B-II protein expression levels
compared with vehicle-treated cells (P<0.01) and further
enhanced the Zoledronate-induced increase in LC3B-II levels
compared with cells treated with Zoledronate only (P<0.01;
Fig. 2A). Furthermore,
accumulation of SQSTM1 was also enhanced in HUVECs following
inhibition of lysosomal activity by chloroquine compared with
vehicle-treated cells (P<0.01), and chloroquine treatment
abolished the inhibitory effect of Zoledronate on SQSTM1 levels
(P<0.01; Fig. 2B). Zoledronate
was therefore demonstrated to induce autophagy through increased
autophagic activity rather than inhibition of lysosome
degradation.
Inhibition of autophagy attenuates
Zoledronate-induced apoptosis in HUVECs
The effect of Zoledronate on the viability or
apoptosis of HUVECs was investigated, due to the intricate
crosstalk between autophagy and apoptosis (24). Zoledronate was demonstrated to
significantly decrease the viability of HUVECs in a dose-dependent
manner compared with untreated control (P<0.01), as demonstrated
in Fig. 3A. Cell viability
following treatment with 100 µM Zoledronate was decreased to
68.4±5.8% compared with untreated control cells (P<0.01;
Fig 3A).
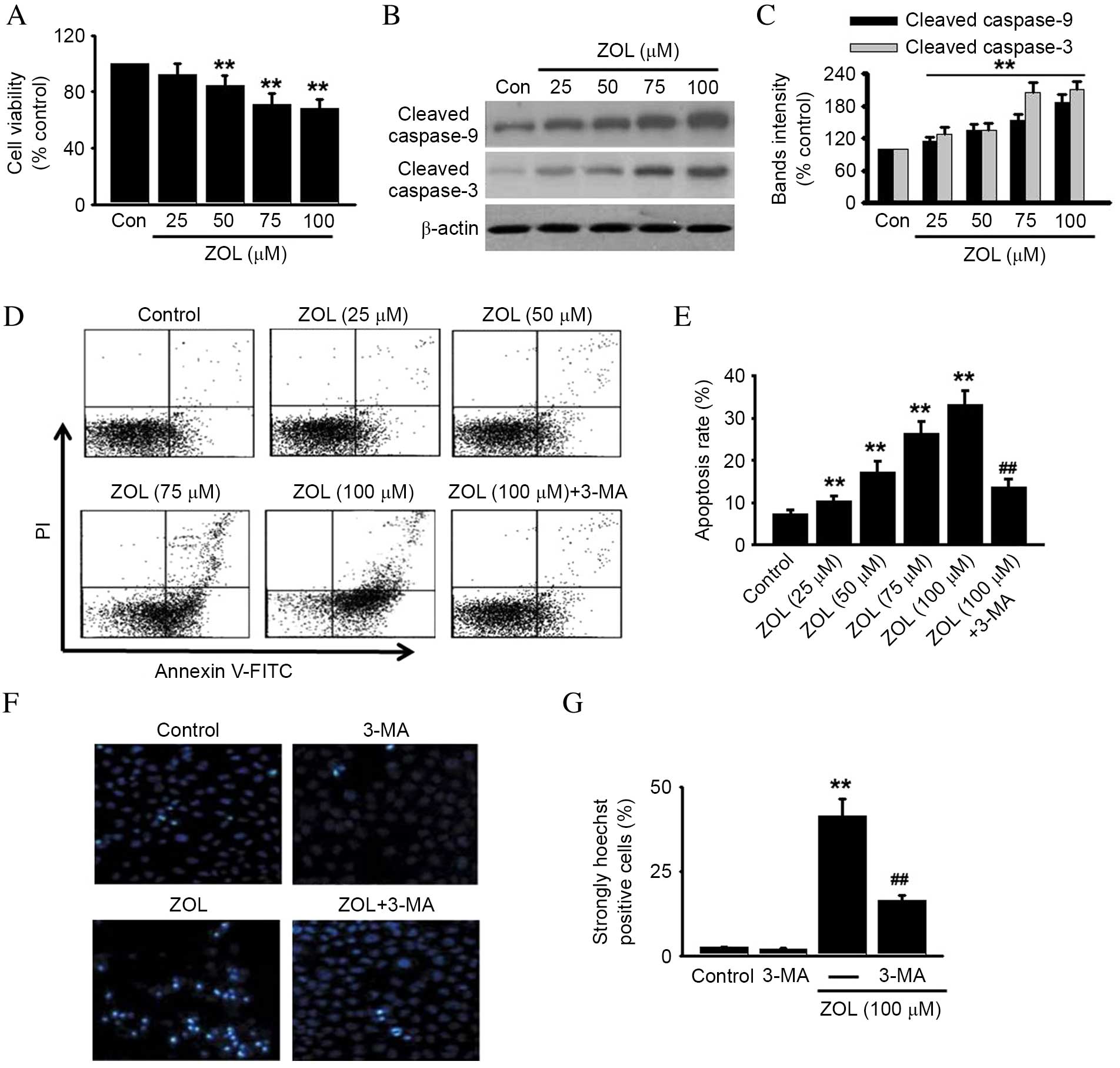 | Figure 3.Inhibition of autophagy protects
against ZOL-induced HUVEC apoptosis. HUVECs were incubated with
various concentrations of ZOL for 48 h, then (A) cell viability was
assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT) assay and (B) cleaved caspase-9 and −3 were detected
by western blot. Representative western blot images are presented.
(C) Densitometric analysis of caspase-9 and cleaved caspase-3
western blots was performed. HUVECs were treated with 5 mM 3-MA
prior to ZOL treatment, then (D) the number of apoptotic cells were
determined by Annexin V-FITC/PI staining using flow cytometry, (E)
followed by quantitative analysis of the percentage of apoptotic
cells. HUVECs treated with 5 mM 3-MA and/or 100 µM ZOL were fixed
and their DNA stained with Hoechst 33258. (F) Confocal images of
nuclear staining are presented and (G) quantification of the number
of strongly Hoechst 33258 positive cells was performed. Data are
presented as the mean ± standard error, n=6. **P<0.01 vs. Con;
##P<0.01 vs. 100 µM ZOL. HUVECs, human umbilical vein
endothelial cells; ZOL, Zoledronate; Con, untreated control; PI,
propidium iodide; FITC, fluorescein isothiocyanate; 3-MA,
3-methyladenine. |
To further determine whether Zoledronate-induced
cell death was due to apoptosis, molecular and cellular changes
associated with apoptosis were analyzed. Increased activation of
caspase signaling was observed, as demonstrated by a significant
increase in the levels of cleaved caspase-9 and caspase-3 following
Zoledronate treatment, compared with untreated cells (P<0.05;
Fig. 3B and C). HUVECs were
subsequently stained with FITC-Annexin-V to detect early apoptotic
cells, and PI to detect late apoptotic/necrotic cells (31). Dose-dependently increased PI and
Annexin-V cell staining was observed in Zoledronate-treated cells
compared with untreated control cells (Fig. 3D), resulting in a significant
dose-dependent increase in the total apoptosis rate (P<0.01;
Fig. 3E). However, the increased
apoptosis induced by 100 µM Zoledronate was significantly inhibited
by 59.2±6.7% following pretreatment with 5 mM 3-MA, an autophagy
inhibitor (P<0.01; Fig. 3D and
E). 3-MA treatment alone exerted no effect on cell apoptosis
(data not shown). Increased nuclear fragmentation in
Zoledronate-treated HUVECs compared with untreated control cells
was also observed by Hoechst 33258 staining (P<0.01; Fig. 3F and G), and was attenuated by
inhibition of autophagy with 3-MA (P<0.01; Fig. 3F and G). Increased autophagy is,
therefore, indicated to be involved in Zoledronate-induced
apoptosis in HUVECs.
Blockade of Beclin-1 inhibits
Zoledronate-induced autophagic death in HUVECs
As Zoledronate was demonstrated to induce an
increase in Beclin-1 expression, it was speculated that the
autophagy induced by Zoledronate was dependent on the Beclin-1
pathway. To examine this hypothesis, Beclin-1 expression was
genetically knocked down in HUVECs prior to Zoledronate exposure,
and changes in cell apoptosis were measured. Transfection
efficiency was confirmed by a significant decrease in Beclin-1
protein expression in siRNA-treated cells (P<0.01; Fig. 4A). Inhibition of Beclin-1
expression decreased the basal LC3B-II protein level compared with
untransfected cells (P<0.05; Fig.
4B), and also abrogated the Zoledronate-induced increase in
LC3B-II protein expression levels when compared with untransfected
cells treated with 100 µM Zoledronate (P<0.01; Fig. 4B). By contrast, inhibition of
Beclin-1 expression increased basal SQSTM1 protein expression
levels compared with untransfected cells (P<0.01; Fig. 4C), and the decreased SQSTM1 protein
expression levels induced by Zoledronate treatment (P<0.01;
Fig. 4C), were partially restored
in Beclin-1 siRNA-transfected cells (P<0.01; Fig. 4C).
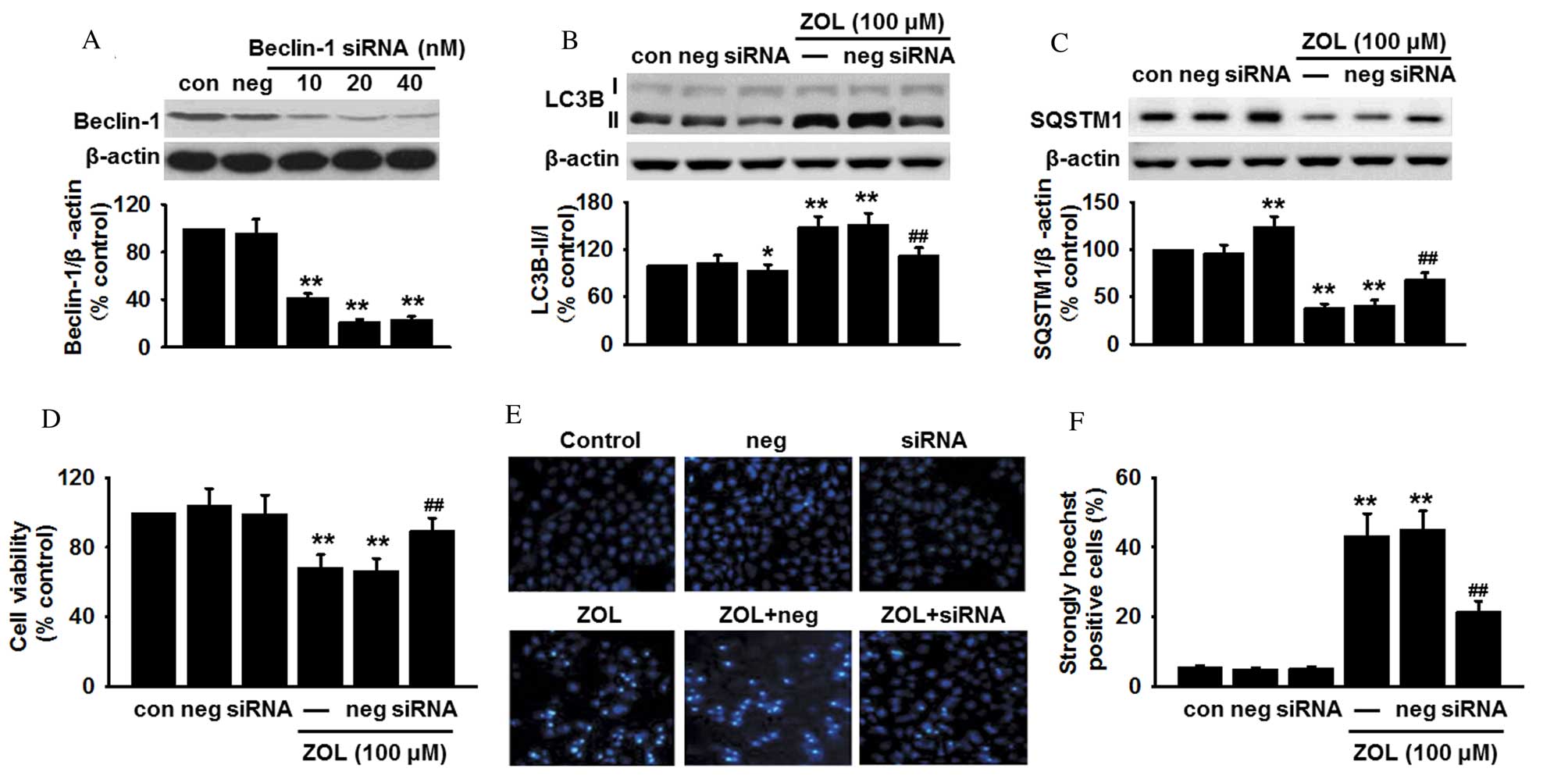 | Figure 4.Absence of Beclin-1 inhibits
ZOL-induced autophagic cell death. (A) HUVECs were transfected with
Beclin-1 siRNA or non-targeting negative control siRNA for 24 h,
and the protein expression level of Beclin-1 determined by western
blot. HUVECs were transfected with Beclin-1 siRNA or non-targeting
negative control siRNA for 24 h, followed by 48 h treatment with
100 µM ZOL. The protein expression levels of (B) LC3B-II and (C)
SQSTM1 were analyzed by western blot, (D) cell viability examined
by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT) assay, and (E) apoptosis detected by Hoechst 33258 staining
and (F) the number of apoptotic cells quantified. Data are
presented as the mean ± standard error, n=6. *P<0.05,
**P<0.01 vs. untransfected control; ##P<0.01 vs.
untransfected cells treated with 100 µM ZOL. HUVECs, human
umbilical vein endothelial cells; siRNA, small interfering RNA;
Con, untransfected control; Neg, negative control siRNA; ZOL,
Zoledronate; LC3B, microtubule-associated proteins 1A/1B light
chain 3B; SQSTM1, sequestome 1. |
The involvement of Beclin-1-mediated autophagy in
HUVEC apoptosis following Zoledronate exposure was subsequently
investigated. Beclin-1 knockdown was revealed to significantly
increase cell viability in HUVECs following 48 h exposure to 100 µM
Zoledronate by MTT assay (P<0.01; Fig. 4D) and Hoechst 33258 staining
(P<0.01; Fig. 4E and F)
compared with untransfected/Zoledronate-treated control cells.
However, knockdown of Beclin-1 did not significantly affect HUVEC
cell viability or Hoechst staining in the absence of Zoledronate
compared with untransfected control cells (Fig. 4D-F). Beclin-1 is, therefore,
suggested to be important for Zoledronate-induced HUVEC
apoptosis.
Discussion
Although numerous studies have suggested that
long-term use of BPs contributes to the development of BP-ONJ
(1,8,11–13),
the underlying molecular mechanism remains poorly understood. To
the best of our knowledge, the present study is the first to
demonstrate the ability of Zoledronate to increase autophagic flux,
thus inducing autophagy in HUVECs. Additionally, inhibition of
autophagy and/or Beclin-1 was demonstrated to reverse the increase
in apoptosis in HUVECs treated with Zoledronate. These findings
suggest a novel mechanism of endothelial cell injury induced by
Zoledronate and provide a potential therapeutic approach for the
treatment of BP-ONJ.
Autophagy is regulated by >30 autophagy specific
genes, including LC3 [a mammalian homologue of autophagy-related
protein 8 (Atg8)], SQSTM1 (also termed p62) and Beclin-1 (a
mammalian homologue of Atg6). During autophagosome formation,
LC3B-I is conjugated with phosphatidylethanolamine at its
C-terminus to form LC3B-II, located in the autophagosome membrane,
which then binds to SQSTM1 to facilitate autophagic degradation in
the lysosome (32,33). LC3B-II, SQSTM1 and Beclin-1 are,
therefore, frequently used as markers of autophagy. The present
study revealed that Zoledronate dose-dependently increased the
expression levels of LC3B-II and Beclin-1, while decreasing the
expression levels of SQSTM1, indicating induction of autophagy.
This supports the previous findings that Zoledronate induced
autophagy in human breast cancer cells (18,34),
cervical cancer cells (21),
salivary adenoid cystic carcinoma cells (22) and human prostate cancer cells
(23).
LC3B-II is localized inside and outside the membrane
of autophagolysosomes (18).
Degradation of the inner LC3B-II in the autophagolysosome can be
prevented by inhibition of autophagolysosome fusion (27). Therefore, activation of autophagy
may not be directly associated with enhanced autophagic activity,
and may be a reduction of autophagolysosome degradation, leading to
accumulation of LC3B-II or feedback stimulation of Beclin-1
(35). By inhibiting lysosomal
activity with chloroquine, the current study demonstrated that
Zoledronate further enhanced the expression of LC3B-II and SQSTM1
in HUVECs with reduced lysosomal activity, indicating that
Zoledronate acts via increased autophagic activity rather than by
reducing autophagolysosome degradation.
Neovascularization at the alveolar ridge following
dental extraction requires endothelial cells during angiogenesis
and vasculogenesis (17). The
majority of clinicians are in agreement that tooth extraction
should be avoided in patients receiving BPs and that BP treatment
should not be resumed until the wound is healed completely
(36). However, clinicians should
be also aware that BPs can accumulate in the bone microenvironment
and that the half-life of BPs can be >10 years (37). Therefore, it is necessary to
further examine the risk factors associated with these drugs.
Previous studies have demonstrated that Zoledronate inhibits the
proliferation, migration and adhesion of endothelial cells,
indicating that Zoledronate may attenuate the viability of
endothelial cells (16,38). The present study demonstrated that
Zoledronate attenuated cell viability and induced apoptosis in
HUVECs, in accordance with a previous study, which reported, that
Zoledronate increased endothelial cell apoptosis and subsequently
reduced angiogenesis in an experimental cervical cancer model
(15).
Autophagy is a complex process, and has a dual
function in apoptosis regulation, depending on the severity and
type of stimuli that induces the autophagy (27,39).
The present study demonstrated that Zoledronate-induced apoptosis
was significantly attenuated following inhibition of autophagy
using 3-MA, indicating that Zoledronate increases endothelial cell
apoptosis, at least partially, by inducing autophagy. Beclin-1 is
important in the regulation of autophagy and the coupling of
autophagy to apoptosis (29).
Previous studies have demonstrated that overexpression of Beclin-1
induces autophagy and inhibits tumor development (19,40).
In the current study, Beclin-1 silencing resulted in inhibition of
HUVEC apoptosis, suggesting that Beclin-1 suppression is important
for the pro-apoptotic effect of Zoledronate.
Notably, the in vitro data in the current
study is not fully representative of the processes in the body,
therefore, further studies are required to verify these effects
in vivo. Furthermore, the mechanisms by which Zoledronate
induces autophagy in endothelial cells, for example the mevalonate
pathway and oxidative stress, should be further investigated.
In summary, the present study demonstrates that
Zoledronate induces HUVEC apoptosis via activation of autophagy,
and that Zoledronate-induced autophagy and apoptosis is inhibited
by siRNA suppression of Beclin-1. These results confirm and enhance
previous findings of the impact of Zoledronate on endothelial
cells, and may guide novel therapeutic strategies to prevent
BP-ONJ.
Acknowledgements
This study was supported by the Nanjing Medical
Science and Technique Development Foundation (grant no. QRX11125)
and the Jiangsu Provincial Clinical Medicine of Science and
Technology project (grant nos. BL2012017 and BL2013005).
References
|
1
|
Bassett CA, Donath A, Macagno F, Preisig
R, Fleisch H and Francis MD: Diphosphonates in the treatment of
myositis ossificans. Lancet. 2:8451969. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Selander KS, Mönkkönen J, Karhukorpi EK,
Härkönen P, Hannuniemi R and Väänänen HK: Characteristics of
clodronate-induced apoptosis in osteoclasts and macrophages. Mol
Pharmacol. 50:1127–1138. 1996.PubMed/NCBI
|
|
3
|
Brown DL and Robbins R: Developments in
the therapeutic applications of bisphosphonates. J Clin Pharmacol.
39:651–660. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tsai SH, Huang PH, Chang WC, Tsai HY, Lin
CP, Leu HB, Wu TC, Chen JW and Lin SJ: Zoledronate inhibits
ischemia-induced neovascularization by impairing the mobilization
and function of endothelial progenitor cells. PloS One.
7:e410652012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ottewell PD, Woodward JK, Lefley DV, Evans
CA, Coleman RE and Holen I: Anticancer mechanisms of doxorubicin
and zoledronic acid in breast cancer tumor growth in bone. Mol
Cancer Ther. 8:2821–2832. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kobayashi Y, Hiraga T, Ueda A, Wang L,
Matsumoto-Nakano M, Hata K, Yatani H and Yoneda T: Zoledronic acid
delays wound healing of the tooth extraction socket, inhibits oral
epithelial cell migration, and promotes proliferation and adhesion
to hydroxyapatite of oral bacteria, without causing osteonecrosis
of the jaw, in mice. J Bone Miner Metab. 28:165–175. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yamashita J, Koi K, Yang DY and McCauley
LK: Effect of zoledronate on oral wound healing in rats. Clin
Cancer Res. 17:1405–1414. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Diel IJ, Bergner R and Grötz KA: Adverse
effects of bisphosphonates: Current issues. J Support Oncol.
5:475–482. 2007.PubMed/NCBI
|
|
9
|
Ruggiero SL, Dodson TB, Assael LA,
Landesberg R, Marx RE and Mehrotra B: American Association of Oral
and Maxillofacial Surgeons: American Association of Oral and
Maxillofacial Surgeons position paper on bisphosphonate-related
osteonecrosis of the jaws-2009 update. J Oral Maxillofac Surg 67 (5
Suppl). 2–12. 2009. View Article : Google Scholar
|
|
10
|
Russell RG: Bisphosphonates: From bench to
bedside. Ann NY Acad Sci. 1068:367–401. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Allen MR and Burr DB: Mandible matrix
necrosis in beagle dogs after 3 years of daily oral bisphosphonate
treatment. J Oral Maxillofac Surg. 66:987–994. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Walter C, Al-Nawas B, Grötz KA, Thomas C,
Thüroff JW, Zinser V, Gamm H, Beck J and Wagner W: Prevalence and
risk factors of bisphosphonate-associated osteonecrosis of the jaw
in prostate cancer patients with advanced disease treated with
zoledronate. Eur Urol. 54:1066–1072. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Reid IR, Bolland MJ and Grey AB: Is
bisphosphonate-associated osteonecrosis of the jaw caused by soft
tissue toxicity? Bone. 41:318–320. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Walter C, Pabst A, Ziebart T, Klein M and
Al-Nawas B: Bisphosphonates affect migration ability and cell
viability of HUVEC, fibroblasts and osteoblasts in vitro. Oral Dis.
17:194–199. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Giraudo E, Inoue M and Hanahan D: An
amino-bisphosphonate targets MMP-9-expressing macrophages and
angiogenesis to impair cervical carcinogenesis. J Clin Invest.
114:623–633. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Santini D, Vincenzi B, Dicuonzo G,
Avvisati G, Massacesi C, Battistoni F, Gavasci M, Rocci L,
Tirindelli MC, Altomare V, et al: Zoledronic acid induces
significant and long-lasting modifications of circulating
angiogenic factors in cancer patients. Clin Cancer Res.
9:2893–2897. 2003.PubMed/NCBI
|
|
17
|
Cetinkaya BO, Keles GC, Ayas B and Gurgor
P: Effects of risedronate on alveolar bone loss and angiogenesis: A
stereologic study in rats. J Periodontol. 79:1950–1961. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Khandelwal VK, Mitrofan LM, Hyttinen JM,
Chaudhari KR, Buccione R, Kaarniranta K, Ravingerová T and
Mönkkönen J: Oxidative stress plays an important role in zoledronic
acid-induced autophagy. Physiol Res. 63:(Suppl 4). S601–S612.
2014.PubMed/NCBI
|
|
19
|
Mizushima N and Levine B: Autophagy in
mammalian development and differentiation. Nat Cell Biol.
12:823–830. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rubinsztein DC, Mariño G and Kroemer G:
Autophagy and aging. Cell. 146:682–695. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang IT, Chou SC and Lin YC: Zoledronic
acid induces apoptosis and autophagy in cervical cancer cells.
Tumour Biol. 35:11913–11920. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ge XY, Yang LQ, Jiang Y, Yang WW, Fu J and
Li SL: Reactive oxygen species and autophagy associated apoptosis
and limitation of clonogenic survival induced by zoledronic acid in
salivary adenoid cystic carcinoma cell line SACC-83. PloS One.
9:e1012072014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lin JF, Lin YC, Lin YH, Tsai TF, Chou KY,
Chen HE and Hwang TI: Zoledronic acid induces autophagic cell death
in human prostate cancer cells. J Urol. 185:1490–1496. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lv XH, Zhao DH, Cai SZ, Luo SY, You T, Xu
BL and Chen K: Autophagy plays a protective role in cell death of
osteoblasts exposure to lead chloride. Toxicol Lett. 239:131–140.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bezzi M, Hasmim M, Bieler G, Dormond O and
Rüegg C: Zoledronate sensitizes endothelial cells to tumor necrosis
factor-induced programmed cell death: Evidence for the suppression
of sustained activation of focal adhesion kinase and protein kinase
B/Akt. J Biol Chem. 278:43603–43614. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Klionsky DJ, Abeliovich H, Agostinis P,
Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA,
Ballabio A, et al: Guidelines for the use and interpretation of
assays for monitoring autophagy in higher eukaryotes. Autophagy.
4:151–175. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ichimura Y and Komatsu M: Selective
degradation of p62 by autophagy. Semin Immunopathol. 32:431–436.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pattingre S, Espert L, Biard-Piechaczyk M
and Codogno P: Regulation of macroautophagy by mTOR and Beclin 1
complexes. Biochimie. 90:313–323. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Esteban-Martinez L and Boya P: Autophagic
flux determination in vivo and ex vivo. Methods. 75:79–86. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Leist M and Jäättelä M: Four deaths and a
funeral: From caspases to alternative mechanisms. Nat Rev Mol Cell
Biol. 2:589–598. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Moscat J and Diaz-Meco MT: p62 at the
crossroads of autophagy, apoptosis, and cancer. Cell.
137:1001–1004. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shvets E, Fass E and Elazar Z: Utilizing
flow cytometry to monitor autophagy in living mammalian cells.
Autophagy. 4:621–628. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wasko BM, Dudakovic A and Hohl RJ:
Bisphosphonates induce autophagy by depleting geranylgeranyl
diphosphate. J Pharmacol Exp Ther. 337:540–546. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Iwai-Kanai E, Yuan H, Huang C, Sayen MR,
Perry-Garza CN, Kim L and Gottlieb RA: A method to measure cardiac
autophagic flux in vivo. Autophagy. 4:322–329. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lacy MQ, Dispenzieri A, Gertz MA, Greipp
PR, Gollbach KL, Hayman SR, Kumar S, Lust JA, Rajkumar SV, Russell
SJ, et al: Mayo clinic consensus statement for the use of
bisphosphonates in multiple myeloma. Mayo Clin Proc. 81:1047–1053.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang EP, Kaban LB, Strewler GJ, Raje N and
Troulis MJ: Incidence of osteonecrosis of the jaw in patients with
multiple myeloma and breast or prostate cancer on intravenous
bisphosphonate therapy. J Oral Maxillofac Surg. 65:1328–1331. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fournier P, Boissier S, Filleur S,
Guglielmi J, Cabon F, Colombel M and Clézardin P: Bisphosphonates
inhibit angiogenesis in vitro and testosterone-stimulated vascular
regrowth in the ventral prostate in castrated rats. Cancer Res.
62:6538–6544. 2002.PubMed/NCBI
|
|
39
|
Zhuang W, Qin Z and Liang Z: The role of
autophagy in sensitizing malignant glioma cells to radiation
therapy. Acta Biochim Biophys Sin (Shanghai). 41:341–351. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|