Introduction
Retinal G protein coupled receptor (RGR)
[Online Mendelian Inheritance in Man (MIM) 600342)] encodes a
putative retinal G-protein coupled receptor, a rhodopsin homologue,
expressed exclusively in the retina (1–3).
RGR is essential for the visual cycle as it is involved in
the production of 11-cis-retinal (4). An abnormal visual cycle affects
visual perception and ultimately leads to ocular disorders
(5). However, the association of
RGR with specific ocular diseases has been rarely reported.
Only a homozygous missense mutation and a heterozygous frameshift
mutation have been reported to be associated with retinitis
pigmentosa and choroidal sclerosis, respectively (5). However, the involvement of RGR
in the pathogenesis of retinitis pigmentosa has not been implicated
in subsequent studies (6,7). The potential role of RGR in
retinal diseases remains to be elucidated. Thus, the present study
aims to systemically evaluate and analyze the potential role and
pathogenicity of variants in RGR. This will be done with
reference to a whole exome sequencing dataset from 820 probands
with different forms of genetic ocular diseases.
Materials and methods
Patients
The present study is part of a project to
investigate genetic defects associated with genetic ocular diseases
using whole exome sequencing. Whole exome sequencing was performed
on samples from 820 probands with different forms of genetic ocular
diseases. All patients were recruited from the clinic of the
Zhongshan Ophthalmic Center (Guangzhou, China). Written informed
consent was obtained from the participants or their guardians,
following the tenets of the Declaration of Helsinki. The present
study was approved by the Institutional Review Board of Zhongshan
Ophthalmic Center.
Sequencing
Whole exome sequencing was performed using a
SureSelect Human All Exon Enrichment kit V4 (Agilent Technologies,
Inc., Santa Clara, CA, USA) or TruSeq Exome Enrichment Kit
(Illumina, Inc., San Diego, CA, USA) as previously described
(8,9). Variants in coding regions and splice
sites in RGR were selected from the whole exome sequencing
data of 820 probands with various genetic ocular diseases. Those
variants with minor allele frequency (MAF) ≤0.01 were further
analyzed by functional prediction using online methods, including
SIFT (sift.jcvi.org/www/SIFT_enst_submit.html) (10), PolyPhen-2 (genetics.bwh.harvard.edu/pph2/) (11), and Berkeley Drosophila Genome
Project (www.fruitfly.org/) (12). The MAF of each variant was obtained
from the public databases, dbSNP (www.ncbi.nlm.nih.gov/projects/SNP/), 1000 Genomes
(www.1000genomes.org/), and the Exome
Variation Server (evs.gs.washington.edu/EVS/). Potential variants of
RGR were further confirmed by Sanger sequencing and
validated in available family members. Primers used for
amplification of fragments were designed using the Primer3 online
tool (bioinfo.ut.ee/primer3-0.4.0/) and are presented in
Table I. The methods used for
amplification, sequencing, and analysis of the target fragments
were as previously described (13). The descriptions of the variants are
consistent with the nomenclature for sequence variations
(www.hgvs.org/mutnomen/) (14).
 | Table I.Primers used for amplification and
sequencing of RGR. |
Table I.
Primers used for amplification and
sequencing of RGR.
| Primer | Forward primer
(5′-3′) | Reverse primer
(5′-3′) | Amplicon (bp) | Annealing temperature
(°C) |
|---|
| RGR-86008695 |
GCAGCATTCAGGAACACACA |
CCCTGCCTCTTATCCTCTCC | 283 | 65–58a |
| RGR-86017741 |
TGCTGACCTGGTTTTCTTGG |
AGGAAGAGACTGACACAGAGGT | 300 | 65–58a |
Results
Following a review of the whole exome dataset of 820
probands with different forms of genetic ocular diseases, a total
of 5 variants of RGR were detected in 6 of the 820 probands.
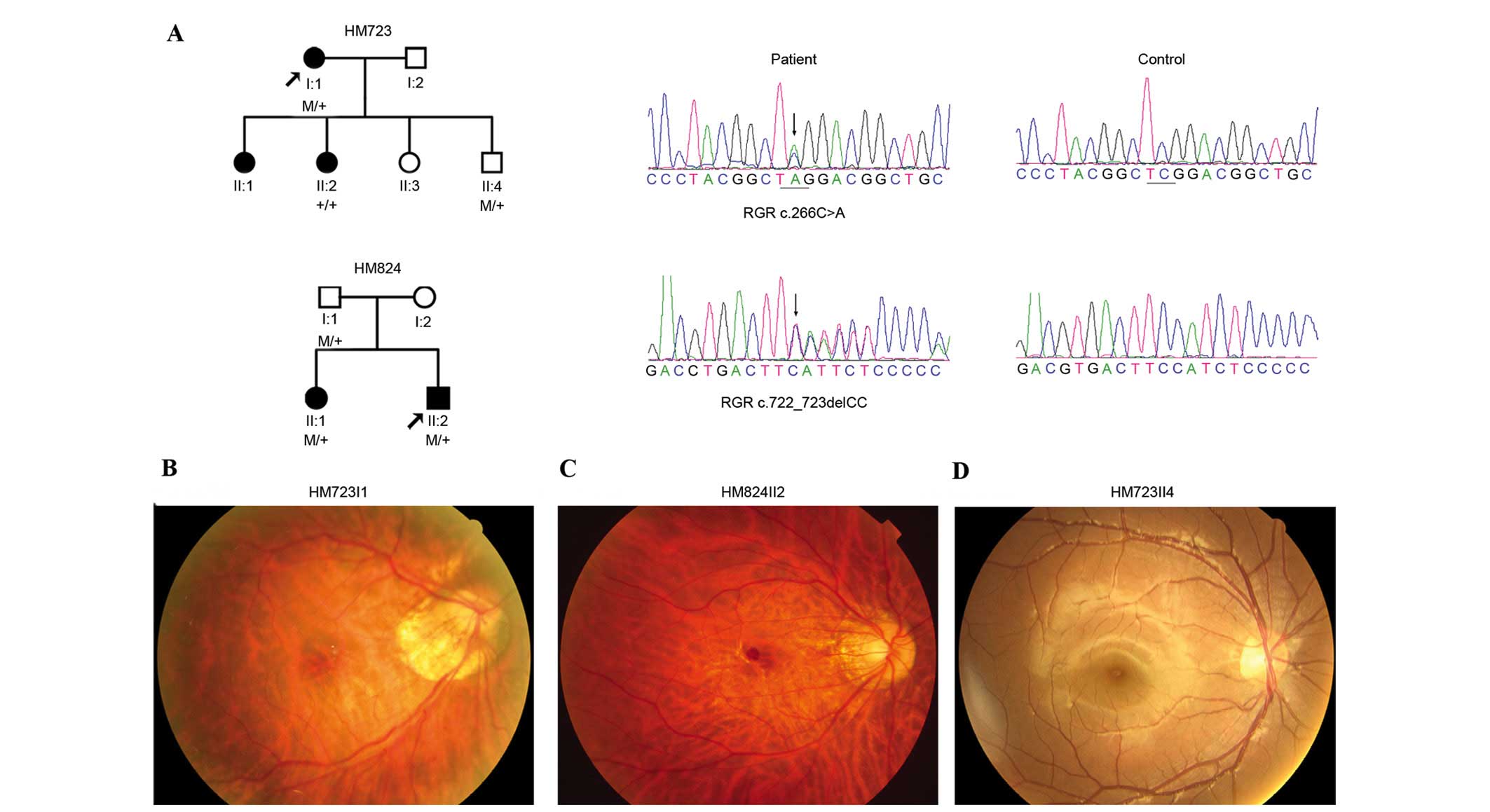
Of the five variants, two were heterozygous truncation variants,
c.266C>A (p.S89*) and c.722_723delCC (p.S242Yfs*29), identified
in two probands with early-onset high myopia (Fig. 1A and Table II). These two variants were
further confirmed by Sanger sequencing (Fig. 1A). Segregation analysis on
available family relatives identified that p.S89* and p.S242Yfs*2
did not co-segregate with high myopia, they were present in the
unaffected relatives but absent in the affected relatives (Fig. 1A). The other three variants were
heterozygous missense variants and identified in four probands, one
with high myopia, one with cone-rod dystrophy, and two with Leber
congenital amaurosis (Table II).
No homozygous or compound heterozygous variants in RGR were
detected.
| Table II.Summary of variants in RGR detected in
probands with different forms of genetic ocular diseases. |
Table II.
Summary of variants in RGR detected in
probands with different forms of genetic ocular diseases.
|
|
|
|
| Variation |
|
| MAF |
|---|
|
|
|
|
|
|
|
|
|
|---|
| Gene | Chromosome | Position | Sample | Nucleotide | Amino acid | Status | SIFT | Poly Phen-2 | 1000G | EVS |
|---|
| RGR | chr10 | 86007503 | HM345, QT371 | c.236G>A | p.R79H | Hetero | D | B | None | None |
| RGR | chr10 | 86007377 | QT1072 | c.110C>T | p.T37I | Hetero | D | B | None | None |
| RGR | chr10 | 86008695 | HM723 | c.266C>A | p.S89* | Hetero | – | – | None | None |
| RGR | chr10 | 86012764 | QT90 | c.522C>G | p.D174E | Hetero | T | D | None | None |
| RGR | chr10 | 86017741 | HM824 | c.722_723delCC | p.S241Yfs*29 | Hetero | – | – | None | None |
The two probands with RGR truncation variants
complained of poor vision at younger than primary school age, but
denied photophobia and night blindness (Table III). Fundus examination
demonstrated tigroid fundus and temporal crescent of optic nerve
head (Fig. 1B and C), which was
consistent with the diagnosis of high myopia. Neither marked
retinal vessel attenuation nor bone corpuscle type of pigmentation
were observed (Fig. 1B and C).
However, additional family members with RGR truncation
variants (HM723II4 and HM824I1) were unaffected individuals without
high myopia (Table III) and did
not have any notable signs of abnormal fundus changes (Fig. 1D).
| Table III.Summary of clinical features in the
families with truncation variants of RGR. |
Table III.
Summary of clinical features in the
families with truncation variants of RGR.
|
| BCA | Refraction (D) | Axial length
(mm) |
|
|---|
|
|
|
|
|
|
|---|
| Case ID | Status | Mutation | Effect | Gender | Age at exam
(years) | First symptom | Right | Left | Right | Left | Right | Left | Fundus |
|---|
| HM723I1 | Affected |
c.[266C>A];[=] | Stopgain | F | 43 | PV | 0.2 | 0.2 | −12.00 | −13.00 | 27.57 | 28.18 | Myopic |
| HM723II2 | Affected | c.[=];[=] | Normal | F | 22 | PV | 0.5 | 0.5 | −7.00 | −6.50 | 26.28 | 26.09 | Normal |
| HM723II4 | Unaffected |
c.[266C>A];[=] | Stopgain | M | 10 | No | 1.2 | 1.0 | −1.00 | −0.50 | 23.18 | 23.24 | Normal |
| HM824II2 | Affected |
c.[722_723delCC];[=] | Frameshift | M | 35 | PV | 0.7 | 0.1 | −15.50 | −18.00 | 31.52 | NAa | Myopic |
| HM824I1 | Unaffected |
c.[722_723delCC];[=] | Frameshift | M | 66 | No | 1.0 | 1.0 | −2.50 | 1.00 | 23.92 | 23.86 | Normal |
Discussion
Based on the whole exome sequencing dataset from 820
probands with different forms of genetic ocular diseases, two
heterozygous truncation variants in RGR were identified in
two probands with high myopia, but these did not co-segregate with
high myopia. The other three variants in RGR were
heterozygous missense variants, and occurred randomly in four
patients with different forms of genetic ocular diseases. No
homozygous or compound heterozygous variants were detected in
RGR.
Only a limited number of RGR variants have
been previously reported (5–7).
Among them, only two have been identified in two families with
either retinitis pigmentosa or choroidal sclerosis (5), a homozygous c.196A>C (p.Ser66Arg)
variant identified in a family with autosomal recessive retinitis
pigmentosa and a heterozygous c.824dupG (p.M275Ifs*83) insertion
identified in a small family with autosomal dominant choroidal
sclerosis (5). Subsequently,
screening of RGR in two independent studies only identified
a number of less likely pathogenic variants and polymorphisms, as
reviewed in Table IV. Of the five
variants detected in the current study, two were heterozygous novel
truncations, p.S89* and p.S242Yfs*29, which presented in two
probands with high myopia. These two variants and the previously
reported heterozygous variant, c.824dupG, were located in exon 3,
exon 6, and exon 7 of RGR, respectively, and have been
predicted to result in an abnormal transcript. They were absent in
the Exome Variants Server and 1000 Genomes databases. However, the
p.S89* and p.S242Yfs*29 variants were also detected in unaffected
family members without any abnormalities of the fundus.
Furthermore, searching of the Exome Variants Server and 1000
Genomes databases revealed a further five truncation variants of
RGR, c.190G>A (p.W47*) in 1/4406 alleles, c.775del1
(p.M260Wfs*43) in 99/12,518 alleles, c.775A>T (p.K259*) in
2/13,006 alleles, c.796_797insCC (p.I267Pfs*37) in 1/12,518
alleles, and c.877C>T (p.R293*) in 1/13,006 alleles. These
findings suggest that heterozygous truncation variants of
RGR are less likely to be pathogenic. Furthermore, it has
been observed that heterozygous missense variants of RGR
have a similar distribution among probands with different forms of
genetic ocular diseases and thus, may not be pathogenic. The
pathogenicity of the homozygous or compound variants of RGR,
remains to be elucidated, as no such variants were detected in the
current study.
| Table IV.Reported variants in RGR. |
Table IV.
Reported variants in RGR.
| First author,
year | Nucleotide | Protein | Status | MAF case | Phenotype in
case | Co-segre
gation | MAF in control | Refs. |
|---|
| Morimura, 1999 |
c.824dupGa | Gly275Ilefs*83 | Hetero | 1/1684 | adRPb | Yes | 0/190 | (5) |
| Morimura, 1999 |
c.196A>Ca | Ser66Arg | Homo | 2/1684 | arRP | Yes | 0/190 | (5) |
| Morimura, 1999 |
IVS5-12A→Gc | Splicing | Hetero | 1/1684 | sRP | NA | 0/190 | (5) |
| Morimura, 1999 |
IVS6+3A→Gc | Splicing | Hetero | 1/1684 | sRP | NA | 0/190 | (5) |
| Morimura, 1999 |
IVS6+5A→Gc | Splicing | Hetero | 4/1684 | sRP | NA | 1/190 | (5) |
| Morimura, 1999 |
GTG→TTGc | Val132Leu | Hetero | 1/1684 | sRP | NA | 0/190 | (5) |
| Morimura, 1999 |
CAC→AACc | His152Asn | Hetero | 1/1684 | sRP | NA | 0/190 | (5) |
| Morimura, 1999 |
GCA→ACAc | Ala234Thr | Hetero | 1/1684 | sRP | NA | 0/190 | (5) |
| Morimura, 1999 |
TCC→TTCc | Ser241Phe | Hetero/Homo | 6/1684 | adRP; sRP | NA | 1/190 | (5) |
| Bernal, 2003 |
TCC→TTCc | Ser241Phe | Hetero | 10/184 | arRP | No | 5/190 | (6) |
| Bernal, 2003 | nt 615
G>Ac | p.= | Hetero | 1/184 | arRP | NA | NA | (6) |
| Bernal, 2003 | IVS6+5
A>G*c | Splicing | Hetero | 1/184 | arRP | No | 0/190 | (6) |
| Ksantini, 2010 |
c.466C>Ac | His156Asn | Hetero/Homo | 3/662 | arRP; sRP | NA | 0/100 | (7)d |
| Ksantini, 2010 |
c.474C>Tc | p.= | NA | 1/184 | sRP | NA | NA | (7) |
| Morimura,1999;
Bernal, 2003 |
IVS5+16C→Te | Intronic | NA | 0.07 | NA | NA | NA | (5,6) |
| Morimura, 1999;
Bernal, 2003 | nt 19
C>Te | p.= | NA | 0.07 | NA | NA | NA | (5,6) |
| Morimura, 1999;
Bernal, 2003 | nt 27
C>Te | p.= | NA | 0.47 | NA | NA | NA | (5,6) |
| Morimura, 1999;
Bernal, 2003 | nt 459
C>Te | p.= | NA | 0.37 | NA | NA | NA | (5,6) |
| Ksantini, 2010 |
c.19C>Te | p.= | NA | 0.03 | NA | NA | NA | (7) |
| Ksantini, 2010 |
c.27T>Ce | p.= | NA | 0.36 | NA | NA | NA | (7) |
| Ksantini, 2010 |
c.-111A>Ge | Non coding | NA | 0.72 | NA | NA | NA | (7) |
| Ksantini, 2010 | c.79 +
59C>Te | Non coding | NA | 0.02 | NA | NA | NA | (7) |
| Ksantini, 2010 | c.642 +
16G>Ae | Non coding | NA | 0.07 | NA | NA | NA | (7) |
| Ksantini, 2010 |
c.*65A>Ge | Non coding | NA | 0.11 | NA | NA | NA | (7) |
| Ksantini, 2010 |
c.*100_101insAe | Non coding | NA | 0.06 | NA | NA | NA | (7) |
In conclusion, the results of the present study
suggest that the potential role of heterozygous truncation of
RGR in ocular diseases remains to be determined. Additional
studies are required to provide further understanding.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. U1201221), the
Natural Science Foundation of Guangdong (grant no. S2013030012978),
and the Fundamental Research Funds of the State Key Laboratory of
Ophthalmology (grant no. 2012PI01).
References
|
1
|
Jiang M, Pandey S and Fong HK: An opsin
homologue in the retina and pigment epithelium. Invest Ophthalmol
Vis Sci. 34:3669–3678. 1993.PubMed/NCBI
|
|
2
|
Shen D, Jiang M, Hao W, Tao L, Salazar M
and Fong HK: A human opsin-related gene that encodes a
retinaldehyde-binding protein. Biochemistry. 33:13117–13125. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Trifunovic D, Karali M, Camposampiero D,
Ponzin D, Banfi S and Marigo V: A high-resolution RNA expression
atlas of retinitis pigmentosa genes in human and mouse retinas.
Invest Ophthalmol Vis Sci. 49:2330–2336. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen P, Lee TD and Fong HK: Interaction of
11-cis-retinol dehydrogenase with the chromophore of retinal g
protein-coupled receptor opsin. J Biol Chem. 276:21098–21104. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Morimura H, Saindelle-Ribeaudeau F, Berson
EL and Dryja TP: Mutations in RGR, encoding a light-sensitive opsin
homologue, in patients with retinitis pigmentosa. Nat Genet.
23:393–394. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bernal S, Calaf M, Garcia-Hoyos M,
Garcia-Sandoval B, Rosell J, Adan A, Ayuso C and Baiget M: Study of
the involvement of the RGR, CRPB1, and CRB1 genes in the
pathogenesis of autosomal recessive retinitis pigmentosa. J Med
Genet. 40:e892003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ksantini M, Sénéchal A, Bocquet B, Meunier
I, Brabet P and Hamel CP: Screening genes of the visual cycle RGR,
RBP1 and RBP3 identifies rare sequence variations. Ophthalmic
Genet. 31:200–204. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang X, Li M, Guo X, Li S, Xiao X, Jia X,
Liu X and Zhang Q: Mutation analysis of seven known
glaucoma-associated genes in Chinese patients with glaucoma. Invest
Ophthalmol Vis Sci. 55:3594–3602. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li J, Jiang D, Xiao X, Li S, Jia X, Sun W,
Guo X and Zhang Q: Evaluation of 12 myopia-associated genes in
Chinese patients with high myopia. Invest Ophthalmol Vis Sci.
56:722–729. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kumar P, Henikoff S and Ng PC: Predicting
the effects of coding non-synonymous variants on protein function
using the SIFT algorithm. Nat Protoc. 4:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Flanagan SE, Patch AM and Ellard S: Using
SIFT and PolyPhen to predict loss-of-function and gain-of-function
mutations. Genet Test Mol Biomarkers. 14:533–537. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Reese MG, Eeckman FH, Kulp D and Haussler
D: Improved splice site detection in Genie. J Comput Biol.
4:311–323. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang D, Li J, Xiao X, Li S, Jia X, Sun W,
Guo X and Zhang Q: Detection of mutations in LRPAP1, CTSH, LEPREL1,
ZNF644, SLC39A5, and SCO2 in 298 families with early-onset high
myopia by exome sequencing. Invest Ophthalmol Vis Sci. 56:339–345.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
den Dunnen JT and Antonarakis SE: Mutation
nomenclature extensions and suggestions to describe complex
mutations: A discussion. Hum Mutat. 15:7–12. 2000. View Article : Google Scholar : PubMed/NCBI
|