Introduction
The preservation of cognitive function during aging
is becoming an increasingly critical public health problem and
emerging as one of the major medical challenges of the 21st
century, particularly in countries with rapidly aging populations
(1,2). Alzheimer's disease, the most frequent
cause of dementia, is an age-related neurodegenerative disease,
which is characterized by amyloid-β (Aβ) peptide deposition
extracellularly and hyperphosphorylated tau protein accumulation
intracellularly as neurofibrillary tangles (3,4).
Although 1–2% of cases of Alzheimer's disease are derived from
pathogenic mutations in amyloid precursor protein (APP) and
presenilin 1/2 genes, the majority of cases are sporadic and tend
to result from pathogenic interactions between multiple factors,
including APP/Aβ aging, genetic variants and apolipoprotein E
genotype (5). Although these
previous investigations indicate notable links between certain
genes and Alzheimer's disease, the exact mechanism underlying the
progression of Alzheimer's disease remains to be fully
elucidated.
Genome-wide approaches have reshaped the landscape
of medical investigations. One of the key mechanisms by which gene
expression is controlled, is examined via a complicated process,
termed transcriptional regulation, which is mediated through
transcription factors. However, the identification of a class of
small non-coding RNAs, termed microRNAs (miRNAs), has augmented the
complexity of transcriptional regulation and opened a novel field
of medical investigations. miRNAs, which consist of 18–24
nucleotides, frequently bind to the 3′-untranslated region (3′-UTR)
of target transcripts and regulate gene expression by degrading the
target mRNA or inhibiting its translation. They are associated with
several biological processes, including development, apoptosis and
proliferation, and their differential expression has been detected
in several disorders, including different types of cancer (6), stroke (7), diabetes (8), and neurological (9), and autoimmune diseases (10,11).
Compared with differential expression analysis, network analysis
may offer an improved method of revealing the correlation between
genes and phenotypes of interest, which are increasingly used in
systems biology. Weighted gene co-expression network analysis
(WGCNA), which was developed by Langfelder and Horvath (12), describes the correlations among
genes and forms gene modules according to their expression values
across microarray samples. However, reports on the association
between miRNAs and Alzheimer's disease using a high throughput
method are limited. For this reason, it is important to identify
and analyze miRNAs, which are likely to be significant in the
expression of disease-associated genes. The aim of the present
study was to identify key genes and biological processes
potentially associated with the progression of Alzheimer's disease,
which may aid development of small, bioactive molecules. The
present study provides important data and theoretical support for
investigations of the physiological and pathological processes of
Alzheimer's disease.
Materials and methods
Microarray dataset
The mRNA expression profile of GSE28146 (13) and the miRNA expression profile of
GSE16759 (14) were downloaded
from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). In GSE28146,
eight control samples, and samples of Alzheimer's disease
comprising seven incipient, eight moderate and seven severe samples
were included. Total RNA was extracted from the
formalin-fixed-paraffin-embedded hippocampus (from the GEO public
database), and hybridized in the GPL570 Affymetrix HGU 133 Plus 2
microarray (Affymetrix, Inc., Santa Clara, CA, USA). For GSE16759,
the miRNA and mRNA expression were profiled for four control
samples and four Alzheimer's disease samples. In the present study,
only those miRNA expression profiles detected via the GPL8757
USC/XJZ Human 0.9 K miRNA-940-v1.0 microarray platform (www.chibi.ubc.ca/Gemma/arrays/showArrayDesign.html?id=680)
were used in the subsequent analysis.
Microarray data analysis
The raw data were converted into a recognizable
format in R software. The Affy (15) package (bioconductor.org/packages/release/bioc/html/affy.html)
was used for the background correction and normalization of the
data. The differentially expressed mRNAs and miRNAs (DEmiRNAs)
between the Alzheimer's disease and control samples were identified
using the Limma (16) package
(bioconductor.org/packages/release/bioc/html/limma.html)
with the following criteria: False discovery rate <0.05 and
|log2 (fold change)|>1.
Weighted gene co-expression network
analysis (WGCNA) of the mRNA microarray
The WGCNA package of R was used for clustering of
the first 5,000 genes with the highest coefficient of variation
[CV; (standard deviation) / (mean value) ×100%)] between
Alzheimer's disease in the mRNA microarray. Age, sex and disease
stage were screened out based on the correlation coefficient and
P-value, which were considered to be the clinical data and gene
modules closely associated with Alzheimer's disease. Gene Ontology
(GO; geneontology.org) and Kyoto Encyclopedia
of Genes and Genomes (KEGG; www.genome.jp/kegg) pathway enrichment analysis were
performed using the Database for Annotation, Visualization and
Integrated Analysis (DAVID; (http://david.abcc.ncifcrf.gov/) in the hub module
(most significant module in the network).
Screening target genes of
DEmiRNAs
The validated target genes of the DEmiRNAs were
obtained using the miRWalk database
(zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2) based on the
combination of miRanda, TargetScan, Pictar and miRbase (17). In addition, the overlap among
target genes, genes in the hub module and the DEGs were identified.
The miRNA-mRNA regulation network was constructed based on the
overlapped genes and their upstream miRNAs, and visualized using
Cytoscape software version 2.6.3 (www.cytoscape.org) (18).
Results
DEGs and DEmiRNAs
A total of 17,559 genes and 90 DEGs were obtained in
the incipient, moderate and severe Alzheimer's disease samples,
respectively, compared with control samples. The numbers of
upregulated and downregulated genes are listed in Table I. In addition, 164 DEmiRNAs, which
contained 74 downregulated and 90 upregulated DEmiRNAs were
identified in the Alzheimer's disease samples.
 | Table I.Numbers of total, upregulated and
downregulated DEGs in the three Alzheimer's disease sample
groups. |
Table I.
Numbers of total, upregulated and
downregulated DEGs in the three Alzheimer's disease sample
groups.
| Sample group | Total DEGs (n) | Upregulated | Downregulated |
|---|
| Incipient | 175 | 53 | 122 |
| Moderate | 59 | 22 | 37 |
| Severe | 90 | 31 | 59 |
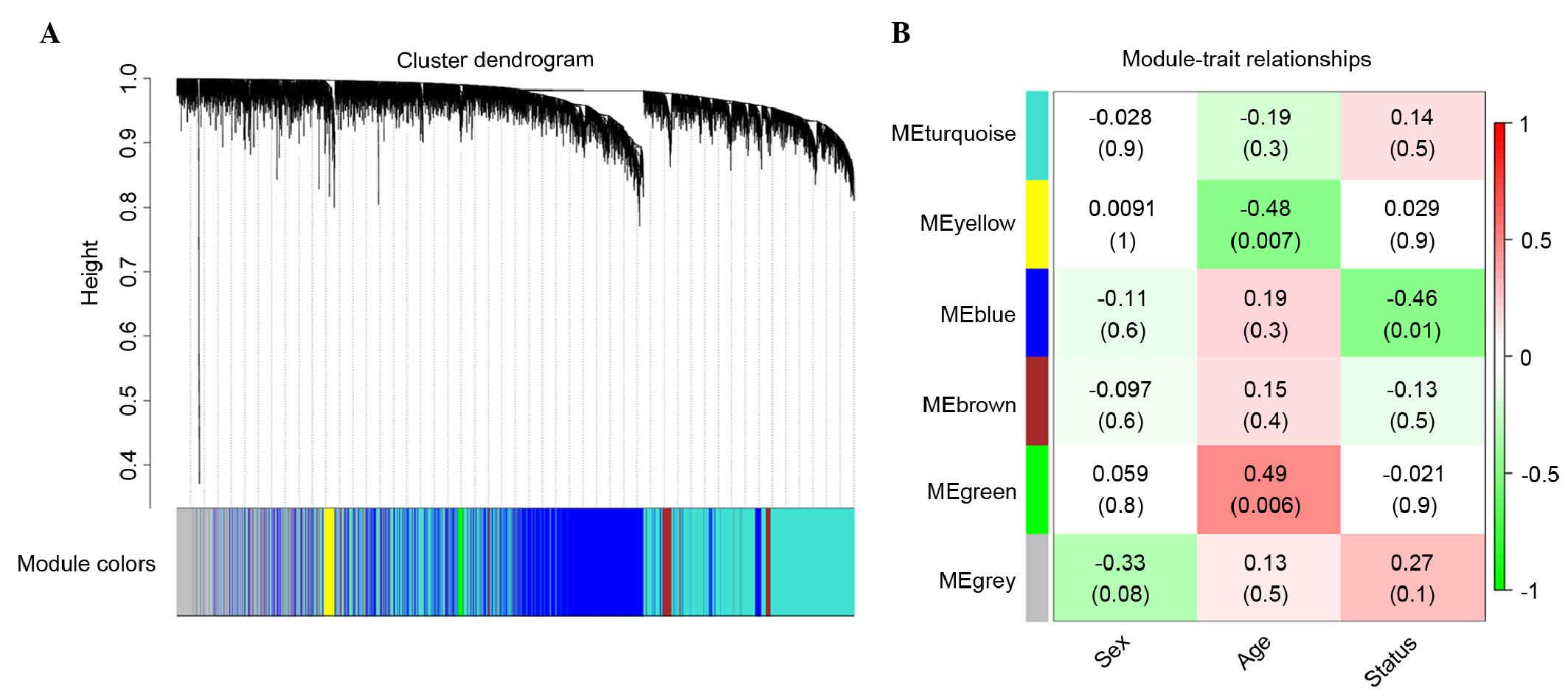
Hub modules of Alzheimer's
disease
Through the WGCNA, the first 5,000 genes with the
highest CV between the control samples and Alzheimer's disease
samples in the mRNA microarray were divided into five gene modules.
The largest of these modules contained 2,200 genes, whereas only 52
genes were included in the smallest module. A total of 787 genes
were not included in any of the five modules. The correlation
coefficients between every gene module, and the age, sex and stage
of Alzheimer's disease were also obtained. The distribution of
genes in the five gene modules and the importance of each gene
module are shown in Fig. 1A and B,
respectively.
As shown in Fig.
1B, the blue module, which contained 1,592 genes, was the most
significant module (correlation coefficient=−0.46; P=0.01)
associated with the stage of Alzheimer's disease. Functional
enrichment analysis indicated that the GO terms and KEGG pathways
associated with the biological processes or diseases of the nervous
system were enriched in the genes of the blue module. The top 10
significant GO terms are shown in Table II. Table III shows the 14 enriched KEGG
pathways.
| Table II.Top 10 significant GO terms enriched
in genes of the blue module. |
Table II.
Top 10 significant GO terms enriched
in genes of the blue module.
| Category | GO term | Number of Genes | P-value |
|---|
| BP | Transmission of nerve
impulse | 64 |
7.00×10−11 |
| BP | Synaptic
transmission | 55 |
1.28×10−9 |
| CC | Mitochondrion | 138 |
4.21×10−9 |
| CC | Synapse | 61 |
6.76×10−9 |
| CC | Synaptic vesicle | 24 |
7.41×10−9 |
| CC | Mitochondrial
part | 87 |
1.02×10−9 |
| CC | Neuron
projection | 59 |
1.06×10−9 |
| CC | Organelle
membrane | 137 |
1.34×10−9 |
| CC | Synapse part | 47 |
1.73×10−8 |
| BP | Generation of
precursor metabolites and energy | 52 |
1.36×10−7 |
| Table III.Enriched Kyoto Encyclopedia of Genes
and Genomes pathways in genes of the blue module. |
Table III.
Enriched Kyoto Encyclopedia of Genes
and Genomes pathways in genes of the blue module.
| Pathway name | Number of genes | P-value |
|---|
| Oxidative
phosphorylation | 26 |
2.24×10−5 |
| Parkinson's
disease | 23 |
3.82×10−4 |
| Huntington's
disease | 28 |
8.26×10−4 |
| Epithelial cell
signaling in Helicobacter pylori infection | 13 |
6.43×10−3 |
|
Glycolysis/Gluconeogenesis | 12 |
6.66×10−3 |
| Alzheimer's
disease | 23 |
8.93×10−3 |
| Vibrio
cholerae infection | 11 | 0.0114 |
| Oocyte meiosis | 17 | 0.0123 |
| Amyotrophic lateral
sclerosis | 10 | 0.0219 |
| Wnt signaling
pathway | 20 | 0.0285 |
| Pathogenic
Escherichia coli infection | 10 | 0.0336 |
| Pyruvate
metabolism | 8 | 0.0354 |
| Long-term
potentiation | 11 | 0.0401 |
| Ubiquitin mediated
proteolysis | 18 | 0.0415 |
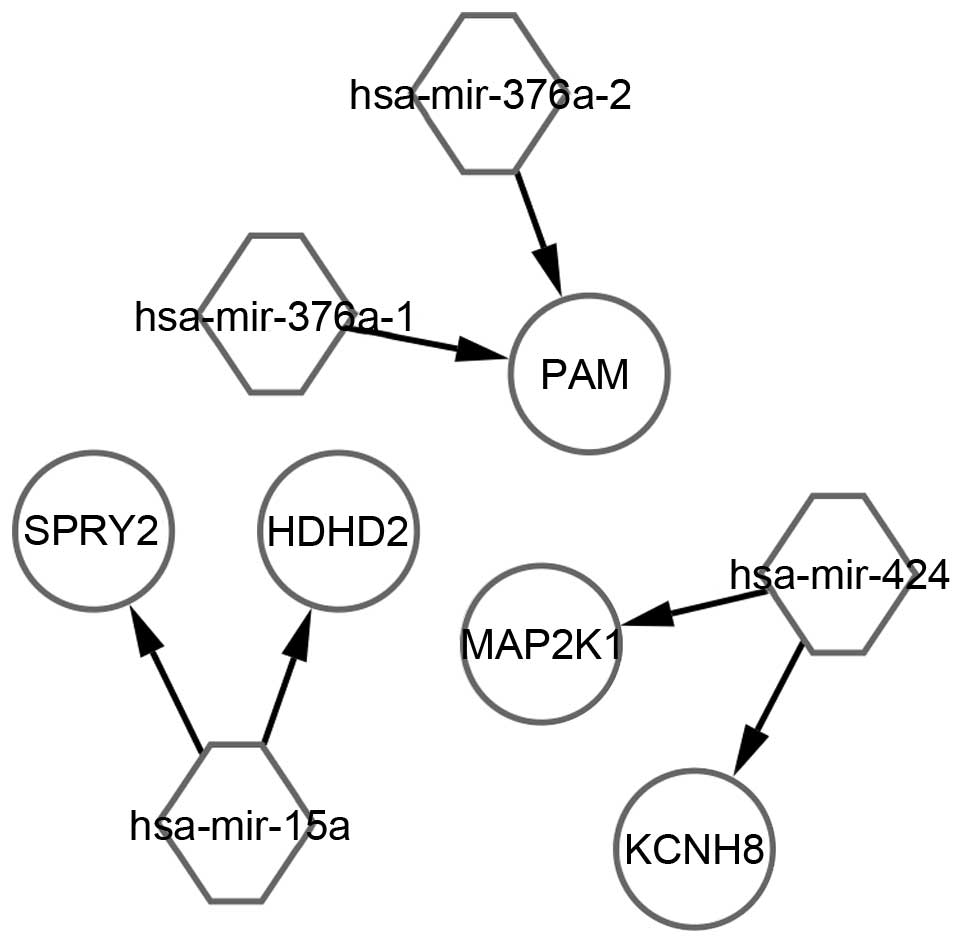
Target genes of DEmiRNAs
In the miRWalk database, 746 non-redundant validated
target genes were identified for the 164 DEmiRNAs. Five overlaps
were obtained, which were regulated by four miRNAs: hsa-mir-376a-2,
hsa-mir-376a-1, hsa-mir-15a and hsa-mir-424. The regulation network
between those overlapped genes and their upstream miRNAs is shown
in Fig. 2.
Discussion
In the present study, microarray technology was used
to identify the therapeutic targets and the associated biological
processes for Alzheimer's disease. Using the combination of
differentially expressed analysis and WGCNA, several potential
targets, including miR-15a and peptidyl-glycine α-amidating
monooxygenase (PAM) were obtained. In addition, several biological
process and KEGG pathways, which were closely associated with
Alzheimer's disease, were further validated.
Sprouty (Spry) proteins contain a carboxyl-terminal
cysteine-rich domain, which is essential for the inhibitory
activity on receptor tyrosine kinase signaling proteins, and they
require growth factors to stimulate translocation of the protein to
membrane ruffles. Over previous years, members of the Spry family
have emerged as critical modulators of growth factor signaling
(19,20). Mammalian spry genes are expressed
in restricted patterns in several adult tissues and in the embryo
during development. They are important in the development of
various organs, including the kidney, lung, testis and tooth
(21–25). At the cellular level, the
overexpression of Spry proteins antagonizes proliferation,
migration and differentiation in response to serum or growth
factors in various cell lines (19,20).
In addition, Gross et al (26) demonstrated that the overexpression
of wild-type Spry2 inhibits neurite formation and expression of
neurofilament light chain, whereas the inhibition of Spry2
accelerates sprouting of multiple neurites. These findings suggest
that Spry2 is potentially involved in neurological disease. The
miR-15a/miR16-1 cluster, located on 13q, is important in regulating
cell proliferation, apoptosis and expression in the brain, heart,
liver, spleen, lung, kidney, small intestine and testis under
physiological conditions (27).
Gao et al (28) found that
miR-15a is important in the regulation of neuronal maturation, and
further confirmed that the overexpression of miR-15a inhibits
dendritic morphogenesis in immature neurons, whereas, a reduction
in miR-15a has the opposite effect. Another study provided evidence
for a potential causal association between the expression of
miR-29a/b-1 and generation of Aβ, with the loss of specific miRNAs
contributing to increased levels of β-secretase 1 (BACE1) and Aβ in
sporadic Alzheimer's disease. Another three miRNAs were also
predicted, which had candidate binding sites within the 3′UTRs of
BACE1, including miR-15a (also found in the present study), miR-9
and miR-19b (29). In the present
study, an important subgroup of patients with Alzheimer's disease
exhibited increased protein expression of Spry2 in the tissue of
the hippocampus, and Spry2 was predicted as an important disease
marker for Alzheimer's disease. The present study also used the
prediction algorithms, miRanda, Targetscan, Pictar and miRbase,
which confirmed that miR-15a had candidate binding sites within the
3′UTRs of Spry2, which indicated miR-15a as a potential major
suppressor of the protein expression of Spry2.
The results of the present study also suggested that
miRNA expression profiles were altered in Alzheimer's disease.
Whether these changes were specific for Alzheimer's disease, or
whether they were a cause or a consequence of the disease process,
and whether these data may be used for diagnosis, remain to be
investigated. Another two miRNAs, miR-376-a1 and miR-376-a2, were
identified as significantly altered, which had candidate binding
sites within the 3′UTRs of PAM, whereas one candidate miRNA-binding
site, miR-424, was found within the 3′ UTRs of mitogen-activated
protein kinase kinase 1 and potassium voltage-gated channel
subfamily H, member 8.
In conclusion, the findings of the present study
suggested a molecular link between miRNA and gene expression
potentially involved in Alzheimer's disease. The functions of these
genes in Alzheimer's disease require further validation through the
use of molecular biology experiments. The present study revealed
potential therapeutic targets and associated biological processes
through the use of differentially expressed gene analysis and
WGCNA, and these findings may assist in the treatment of
Alzheimer's disease.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant nos. 81202739, 81473766 and
81173337) and the Key Project of Tianjin Natural Science Fund
(grant no. 16JCZDJC36200).
References
|
1
|
Sosa AL, Albanese E, Stephan BC, Dewey M,
Acosta D, Ferri CP, Guerra M, Huang Y, Jacob KS, Jiménez-Velázquez
IZ, et al: Prevalence, distribution, and impact of mild cognitive
impairment in Latin America, China and India: A 10/66
population-based study. PLoS Med. 9:e10011702012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ashby-Mitchell K, Jagger C, Fouweather T
and Anstey KJ: Life expectancy with and without cognitive
impairment in seven Latin American and Caribbean countries. PloS
One. 10:e01218672015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Price DL, Tanzi RE, Borchelt DR and
Sisodia SS: Alzheimer's disease: Genetic studies and transgenic
models. Annu Rev Genet. 32:461–493. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Barnes DE and Yaffe K: The projected
effect of risk factor reduction on Alzheimer's disease prevalence.
Lancet Neurol. 10:819–828. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huang Y and Mucke L: Alzheimer mechanisms
and therapeutic strategies. Cell. 148:1204–1222. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gregory RI and Shiekhattar R: MicroRNA
biogenesis and cancer. Cancer Res. 65:3509–3512. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tan JR, Tan KS, Koo YX, Yong FL, Wang CW,
Armugam A and Jeyaseelan K: Blood microRNAs in low or no risk
ischemic stroke patients. Int J Mol Sci. 14:2072–2084. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Balasubramanyam M, Aravind S,
Gokulakrishnan K, Prabu P, Sathishkumar C, Ranjani H and Mohan V:
Impaired miR-146a expression links subclinical inflammation and
insulin resistance in Type 2 diabetes. Mol Cell Biochem.
351:197–205. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Minones-Moyano E, Porta S, Escaramis G,
Rabionet R, Iraola S, Kagerbauer B, Espinosa-Parrilla Y, Ferrer I,
Estivill X and Martí E: MicroRNA profiling of parkinson's disease
brains identifies early downregulation of miR-34b/c which modulate
mitochondrial function. Hum Mol Genet. 20:3067–3078. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Argyropoulos C, Wang K, McClarty S, Huang
D, Bernardo J, Ellis D, Orchard T, Galas D and Johnson J: Urinary
microRNA profiling in the nephropathy of type 1 diabetes. PLoS One.
8:e546622013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Salas-Pérez F, Codner E, Valencia E,
Pizarro C, Carrasco E and Pérez-Bravo F: MicroRNAs miR-21a and
miR-93 are down regulated in peripheral blood mononuclear cells
(PBMCs) from patients with type 1 diabetes. Immunobiology.
218:733–737. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Blalock EM, Buechel HM, Popovic J, Geddes
JW and Landfield PW: Microarray analyses of laser-captured
hippocampus reveal distinct gray and white matter signatures
associated with incipient Alzheimer's disease. J Chem Neuroanat.
42:118–126. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nunez-Iglesias J, Liu CC, Morgan TE, Finch
CE and Zhou XJ: Joint genome-wide profiling of miRNA and mRNA
expression in Alzheimer's disease cortex reveals altered miRNA
regulation. PLoS One. 5:e88982010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: Affy-analysis of affymetrix genechip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: Limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dweep H, Sticht C, Pandey P and Gretz N:
miRWalk-database: Prediction of possible miRNA binding sites by
‘walking’ the genes of three genomes. J Biomed Inform. 44:839–847.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mason JM, Morrison DJ, Basson MA and Licht
JD: Sprouty proteins: Multifaceted negative-feedback regulators of
receptor tyrosine kinase signaling. Trends Cell Biol. 16:45–54.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim HJ and Bar-Sagi D: Modulation of
signalling by Sprouty: A developing story. Nat Rev Mol Cell Biol.
5:441–450. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mailleux AA, Tefft D, Ndiaye D, Itoh N,
Thiery JP, Warburton D and Bellusci S: Evidence that SPROUTY2
functions as an inhibitor of mouse embryonic lung growth and
morphogenesis. Mech Dev. 102:81–94. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gross I, Morrison DJ, Hyink DP, Georgas K,
English MA, Mericskay M, Hosono S, Sassoon D, Wilson PD, Little M
and Licht JD: The receptor tyrosine kinase regulator sprouty1 is a
target of the tumor suppressor WT1 and important for kidney
development. J Biol Chem. 278:41420–41430. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Basson MA, Akbulut S, Watson-Johnson J,
Simon R, Carroll TJ, Shakya R, Gross I, Martin GR, Lufkin T,
McMahon AP, et al: Sprouty1 is a critical regulator of
GDNF/RET-mediated kidney induction. Dev Cell. 8:229–239. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Klein OD, Minowada G, Peterkova R, Kangas
A, Yu BD, Lesot H, Peterka M, Jernvall J and Martin GR: Sprouty
genes control diastema tooth development via bidirectional
antagonism of epithelial-mesenchymal FGF signaling. Dev Cell.
11:181–190. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Minowada G, Jarvis LA, Chi CL, Neubüser A,
Sun X, Hacohen N, Krasnow MA and Martin GR: Vertebrate sprouty
genes are induced by FGF signaling and can cause chondrodysplasia
when overexpressed. Development. 126:4465–4475. 1999.PubMed/NCBI
|
|
26
|
Gross I, Armant O, Benosman S, de Aguilar
JL, Freud JN, Kedinger M, Licht JD, Gaiddon C and Loeffler JP:
Sprouty2 inhibits BDNF-induced signaling and modulates neuronal
differentiation and survival. Cell Death Differ. 14:1802–1812.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yue J and Tigyi G: Conservation of
miR-15a/16-1 and miR-15b/16-2 clusters. Mamm Genome. 21:88–94.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gao Y, Su J, Guo W, Polich ED, Magyar DP,
Xing Y, Li H, Smrt RD, Chang Q and Zhao X: Inhibition of miR-15a
promotes BDNF expression and rescues dendritic maturation deficits
in MeCP2-deficient neurons. Stem Cells. 33:1618–1629. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hebert SS, Horré K, Nicolaï L,
Papadopoulou AS, Mandemakers W, Silahtaroglu AN, Kauppinen S,
Delacourte A and De Strooper B: Loss of microRNA cluster
miR-29a/b-1 in sporadic Alzheimer's disease correlates with
increased BACE1/beta-secretase expression. Proc Natl Acad Sci USA.
105:6415–6420. 2008. View Article : Google Scholar : PubMed/NCBI
|