Introduction
Primary ciliary dyskinesia (PCD) is a rare genetic
disorder of ciliary function and affects ~1/20,000 live births
(1/12,500–1/30,000) (1). Cilia
serve critical roles throughout the human body and at all stages of
life, including during left-right patterning of embryonic organs,
the clearing of mucus and dirt from the respiratory tract, and the
proper movement of sperm or ova; therefore, symptoms of PCD are
diverse and include situs inversus, chronic oto-rhino-pulmonary
infection and infertility. A large number (>200) of genes code
ciliary components, and the symptoms of PCD can vary between
patients. This heterogeneity makes the diagnosis of PCD
challenging, particularly when heterotaxia is absent and other
symptoms are mild (2). This
challenge is a problem, due to the fact that the later PCD is
diagnosed, the worse the prognosis is (3). There is no gold-standard for
diagnosis of PCD, and a comprehensive approach using both
structural and functional analysis of the cilia and genetic
analysis of causative genes is required. In the current study, a
case of PCD in which the patient had been treated for ‘intractable
atelectasis with unknown origin’ for nine years is reported. The
present study highlighted the benefits of diagnostic strategies
that include ultrastructural analysis accompanied by extended
genetic analysis involving whole-exome sequencing.
Patients and methods
Chest computed tomography (CT) images were acquired
by an Aquillion CT scanner (Toshiba Medical Systems Corporation,
Otawara, Japan), with automatic exposure control (SD:12) at 120 kVp
with 5 mm section thicknesses. Nasal mucosa was obtained for
electron microscopy. Under local anesthesia, a small amount of
nasal mucosa was extracted from the patient's inferior turbinate.
According to the methodology of Rubin (4), greater than 100 cilia from the
patient were examined via electron microscopy (JEM-1011; JEOL,
Tokyo, Japan). Nasal mucosa was also extracted from a subject with
no nasal diseases and served as a normal control group.
Genetic analysis was approved by the Mie University
University Ethics Committee (no. 1363), and written informed
consent was obtained from the proband and each parent. Genomic DNA
was extracted from peripheral blood samples taken from the forearm
of each participant. Subsequently, known hot spots were sequenced
in two candidate genes, DNAH5 and DNAI1, due to the
fact that a previous study observed mutations in DNAH5 or
DNAI1 in approximately a third of all patients with PCD
(5).
For the whole-exome sequencing, proband DNA was
amplified with the Ion AmpliSeq™ Exome RDY Kit (Life Technologies;
Thermo Fisher Scientific, Inc., Waltham, MA, USA), which targets
more than 97% of human consensus coding sequences. After quality
control thaws were performed with the Bioanalyzer High Sensitivity
Chip (Agilent Technologies, Inc., Santa Clara, CA, USA) and
emulsion polymerase chain reaction (PCR; Ion PI™ Hi-Q™ OT2 200 kit;
Life Technologies; Thermo Fisher Scientific, Inc.), samples were
sequenced with a Proton PI chip version 3 and the Ion Proton
semiconductor sequencer system (Life Technologies; Thermo Fisher
Scientific, Inc.). Base calling, pre-processing of the reads, short
read alignment and variant calling were performed with the Torrent
Suite, the Torrent Variant Caller (version 4.6; Thermo Fisher
Scientific, Inc.), and the default parameters recommended for the
Ampliseq Exome panel (low stringency calling of germline variants,
version, april 2014). Variant annotation was performed with Ion
Reporter, version 4.6 (Life Technologies; Thermo Fisher Scientific,
Inc.) and was data integrated from a variety of public
databases.
Exome variant analysis was performed by filtering
the whole variant list according to three criteria: i) Consistent
autosomal recessive inheritance patterns, ii) novelty in comparison
to human polymorphism databases [including the 1000 Genomes
(http://www.1000genomes.org/) and dbSNP
(http://www.ncbi.nlm.nih.gov/projects/SNP/)], and iii)
functional significance. These analyses required the presence of at
least one homozygous or two heterozygous changes occurring with an
estimated frequency of <0.01.
Variants were validated via PCR and Sanger
sequencing with the 3500 Series Genetic Analyzer (Thermo Fisher
Scientific, Inc.). These tests were performed according to standard
protocols specifically adapted to preclude technical artifacts and
test for segregation. The primers used for the amplification were
as follows: DNAH5 exon 36 F, 5′-CTTGTGTGCGTTTCATGCCA-3′;
DNAH5 exon 36 R, 5′-CTGCAACCGAGAGAACTGGT-3′; DNAH5
exon 54 F, 5′-GATGATAACGGTGTTGGGGGAT-3′, DNAH5 exon 54 R,
5′-GTAGCCCCGGAAAGGAGTAAAT-3′.
MutationTaster (http://mutationtaster.org/) and Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/)
analyses were conducted in order to predict the impact of variants
in silico.
Results
Clinical information
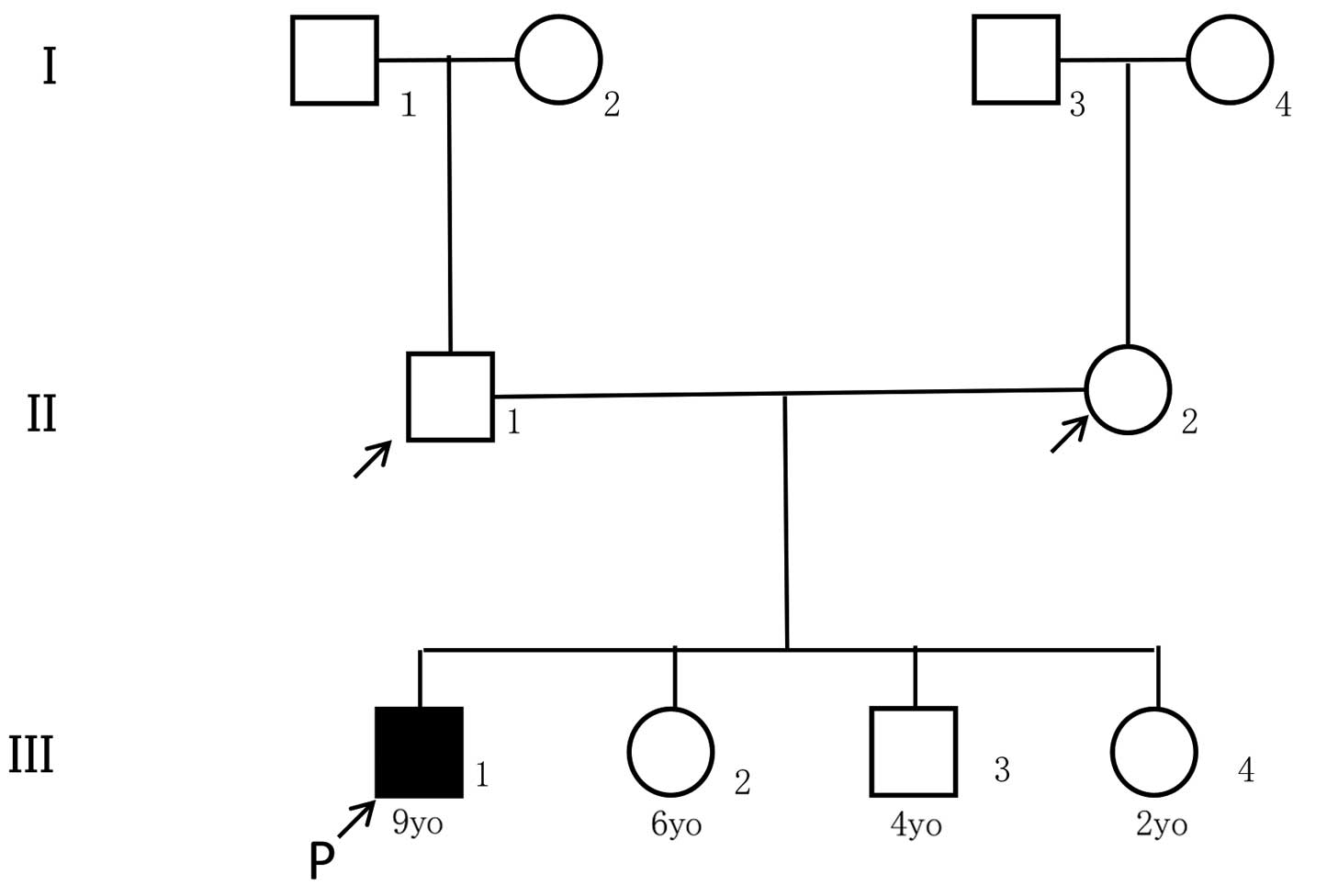
The proband was a nine-year-old boy who was the
offspring from non-consanguineous Japanese parents. His close
relatives, specifically his parents, grandparents, two younger
sisters and one younger brother, did not have a history of
significant respiratory illness (Fig.
1). The first year of his life was uneventful, except that he
experienced mild dyspnea and required low dose oxygen from the
second to fourth days after birth. At one year of age, he was
hospitalized for five days with a diagnosis of asthmatic
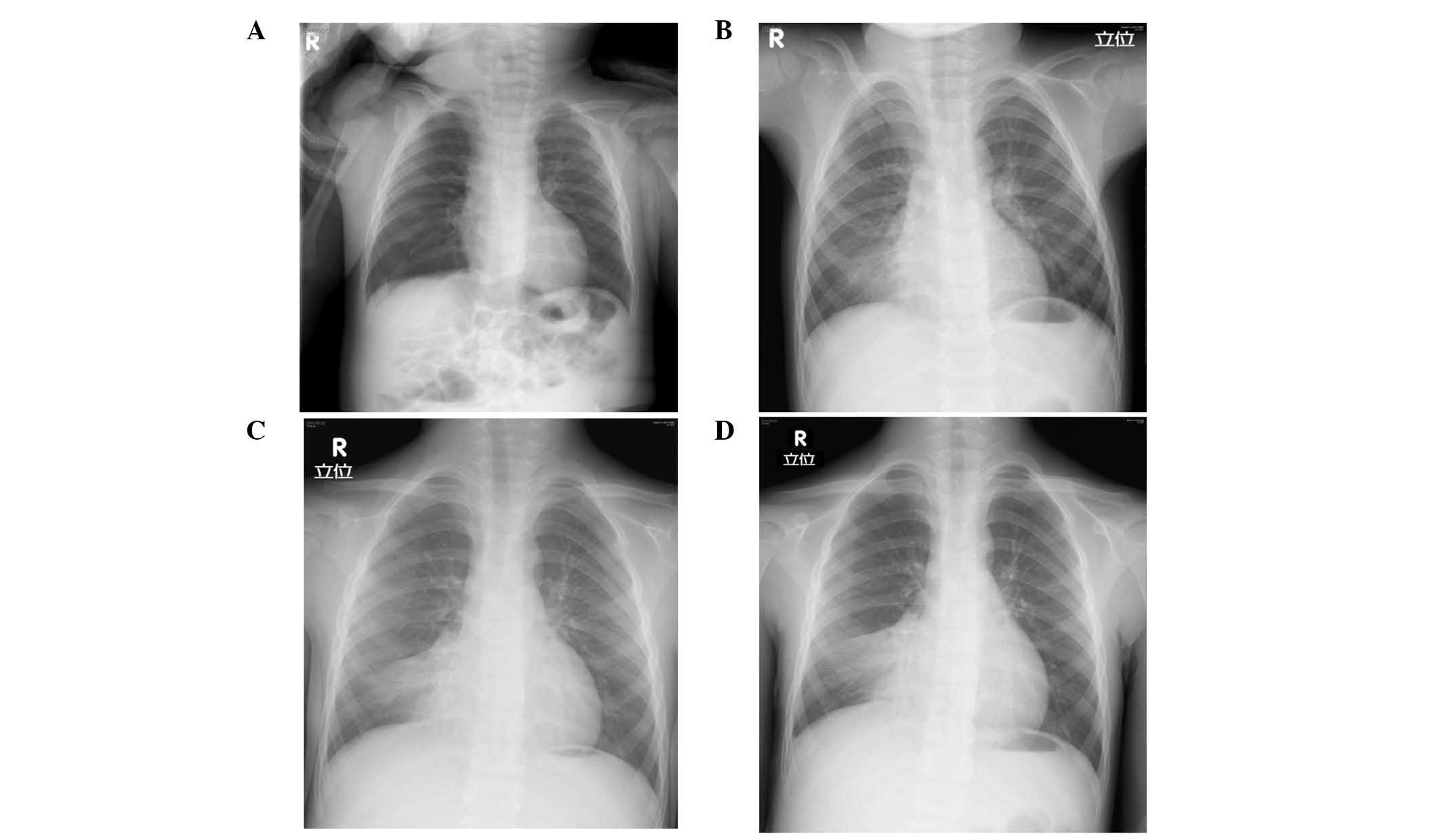
bronchitis, with no significant lung infiltrations observed by
X-ray (Fig. 2A). Since then, the
patient experienced chronic nasal discharge and productive cough
along with multiple episodes of sinusitis and/or otitis media.
Atelectasis of the right lower lobe was first noted on a chest
X-ray at three years of age, when he was admitted for acute
pneumonia (Fig. 2B). Despite
intense physiotherapy including positive expiratory pressure
therapy or high frequency chest wall oscillation, the atelectasis
remained unresolved (Fig. 2C and
D). He was also diagnosed with asthma due to a frequent cough,
leading to administration of inhaled corticosteroids and other
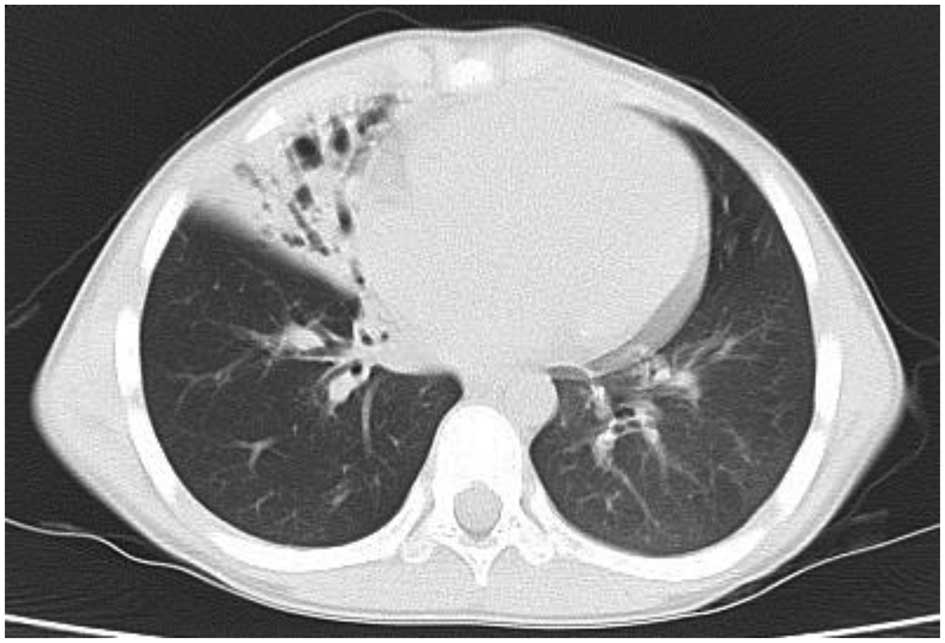
asthma-specific therapies. At nine years of age, bronchiectasis was
observed in the same lobe of the right lung by CT scanning
(Fig. 3). Respiratory function
tests at that point demonstrated mild airway obstruction with
forced vital capacity of 1.64 l (95.3% predicted), forced
expiratory volume1.0 1.18% (80.1% predicted) and maximum
midexpiratory flow 0.73 l/sec (37.2% predicted). All standard
screening tests for immunodeficiency including serum immunoglobulin
levels and T/B lymphocyte counts were normal; consequently, PCD was
suspected, and the patient was referred to the Department of
Otorhinolaryngology at our hospital.
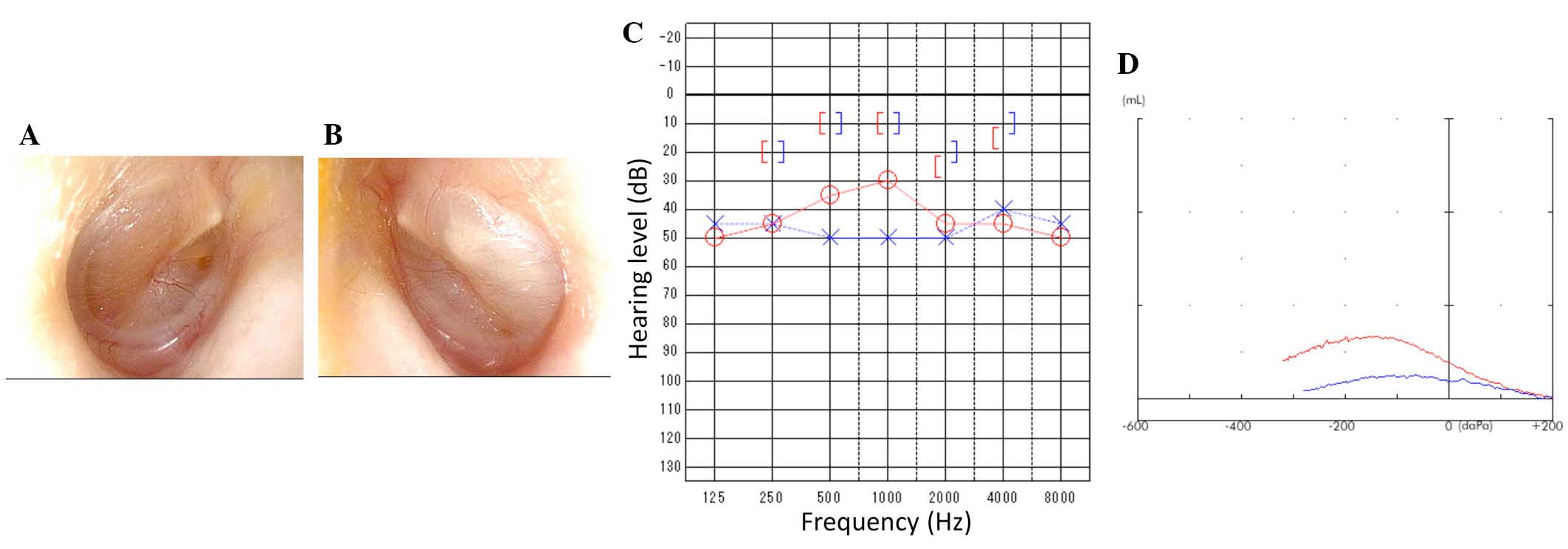
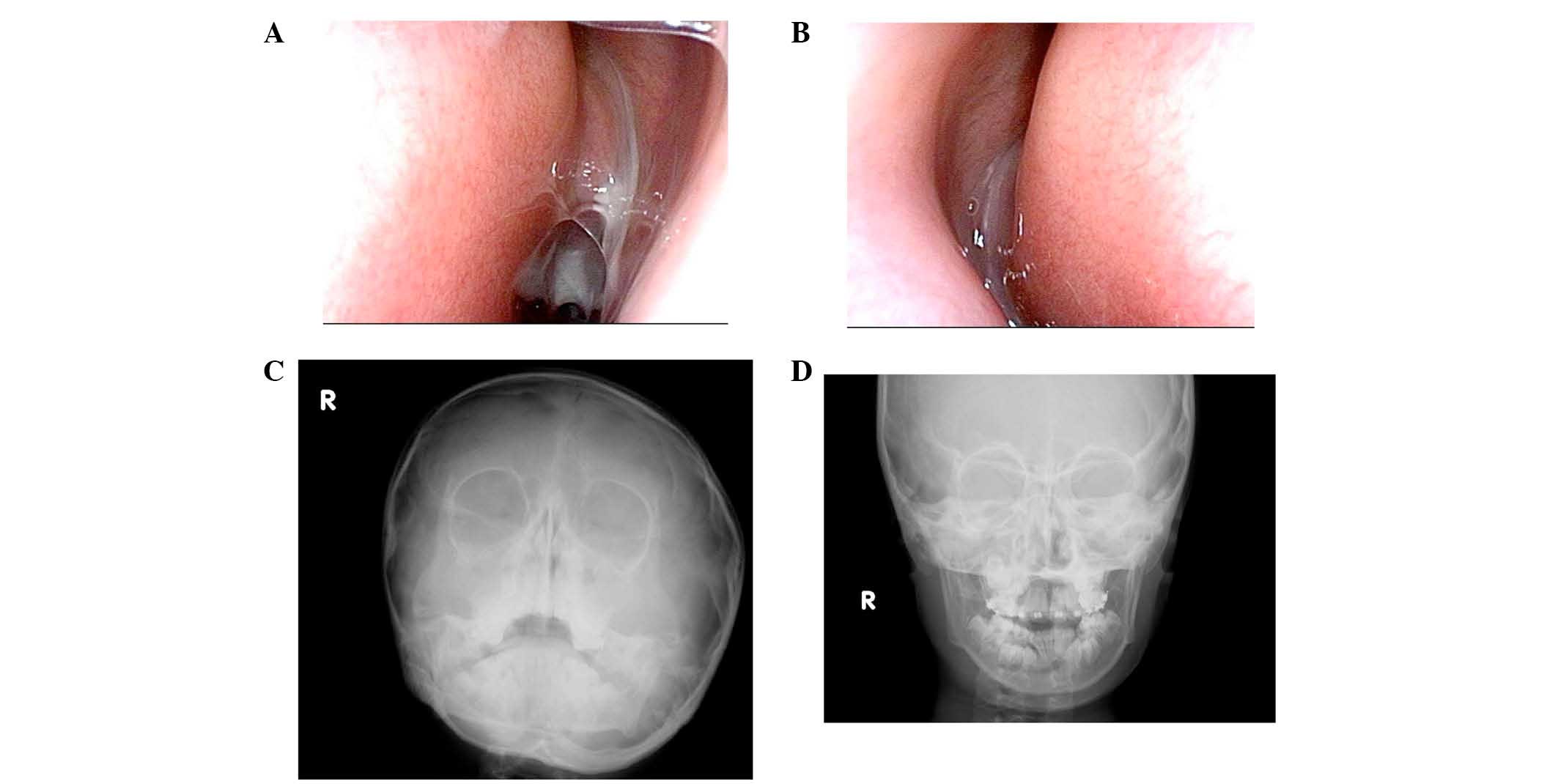
Otological examination revealed that the light
reflex was missing from the right eardrum (Fig. 4A). Additionally, for the left
eardrum, the posterosuperior quadrant was bulging, and the light
reflex was missing (Fig. 4B).
Pure-tone audiometry revealed bilateral conductive hearing loss
with a hearing level of 35 dB in the right and 50 dB in the left
(Fig. 4C). Tympanograms were type
B (Fig. 4D) bilaterally; these
observations were compatible with otitis media with effusion.
Rhinological evaluation demonstrated that the nasal cavities were
filled bilaterally with mucopurulent nasal secretions (Fig. 5A and B). Nasal X-ray revealed
soft-tissue density bilaterally in the maxillary sinuses,
suggesting chronic sinusitis and agenesis of the frontal sinuses
(Fig. 5C and D). Nasal and exhaled
nitric oxide (NO) was measured via an ANALYZER CLD 88®
according to American Thoracic Society/European Respiratory Society
recommendations (6); the
fractional concentration of exhaled NO (FeNO) and nasal NO values
were 10.0 and 0.2 ppb, respectively (data not shown). This low
value of nasal NO was indicative of PCD (7).
Electron microscopy
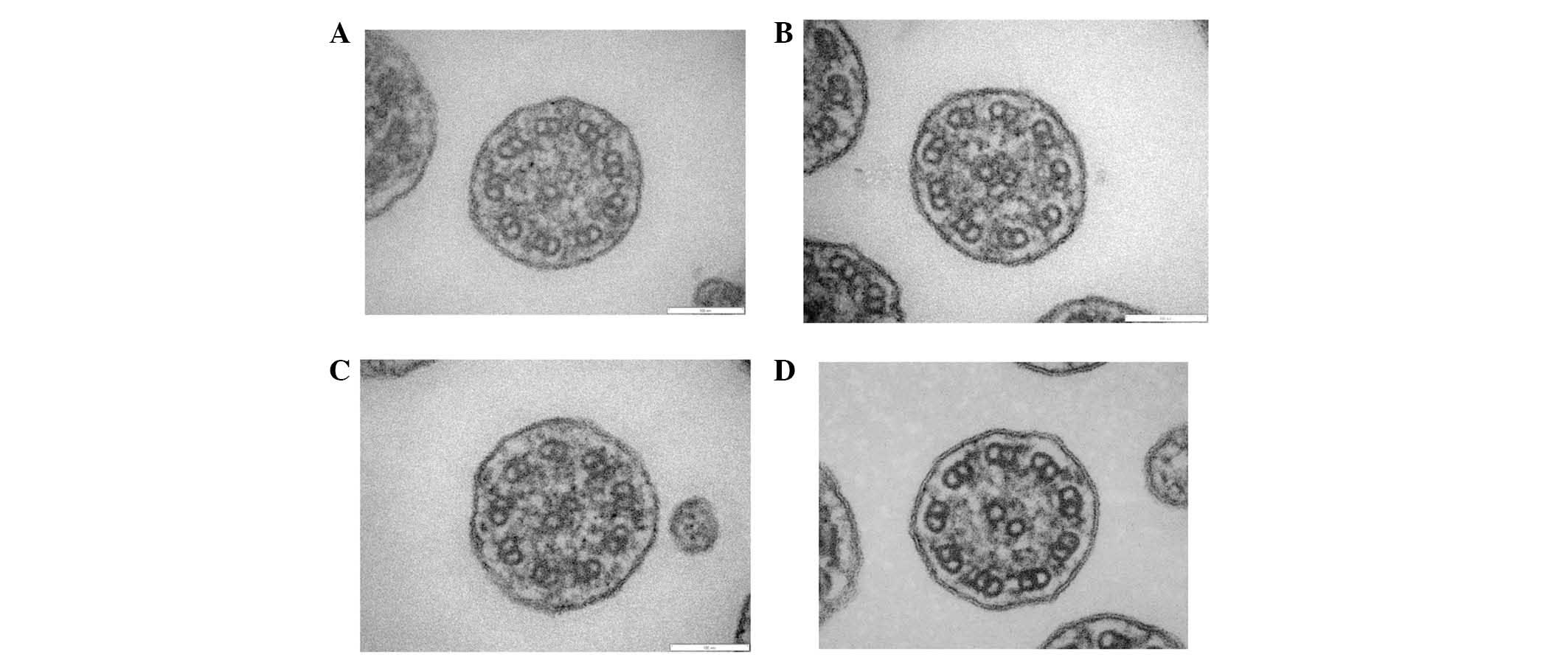
Analysis of the specimen collected from the patient
demonstrated shortened outer dynein arms (Fig. 6A-C); and this observation was
compatible with PCD. In the normal control group (Fig. 6D), both the outer and inner dynein
arms were observed.
Genetic analysis
No causative mutations were identified via
conventional Sanger-based analyses of exons 34, 50, 63, 76 and 77
of DNAH5 (8) and exons 1,
13, 16, and 17 of DNAI1 (9). Based on these observations,
whole-exome sequencing was conducted.
The whole-exome analysis of the proband genomic DNA
identified two novel compound heterozygous mutations in
DNAH5: NM_001369.2:c.5983C>T, p.Arg1995X in exon 36; and
NM_001369.2:c.9101delG, p.Gly3034ValfsX22 in exon 54. PolyPhen-2
analysis indicated that the p.Gly3034ValfsX22 mutation of
DNAH5 was likely to be functionally damaging, with a score
of 1.000. MutationTaster predicted that each of these DNAH5
mutations would cause nonsense-mediated mRNA decay.
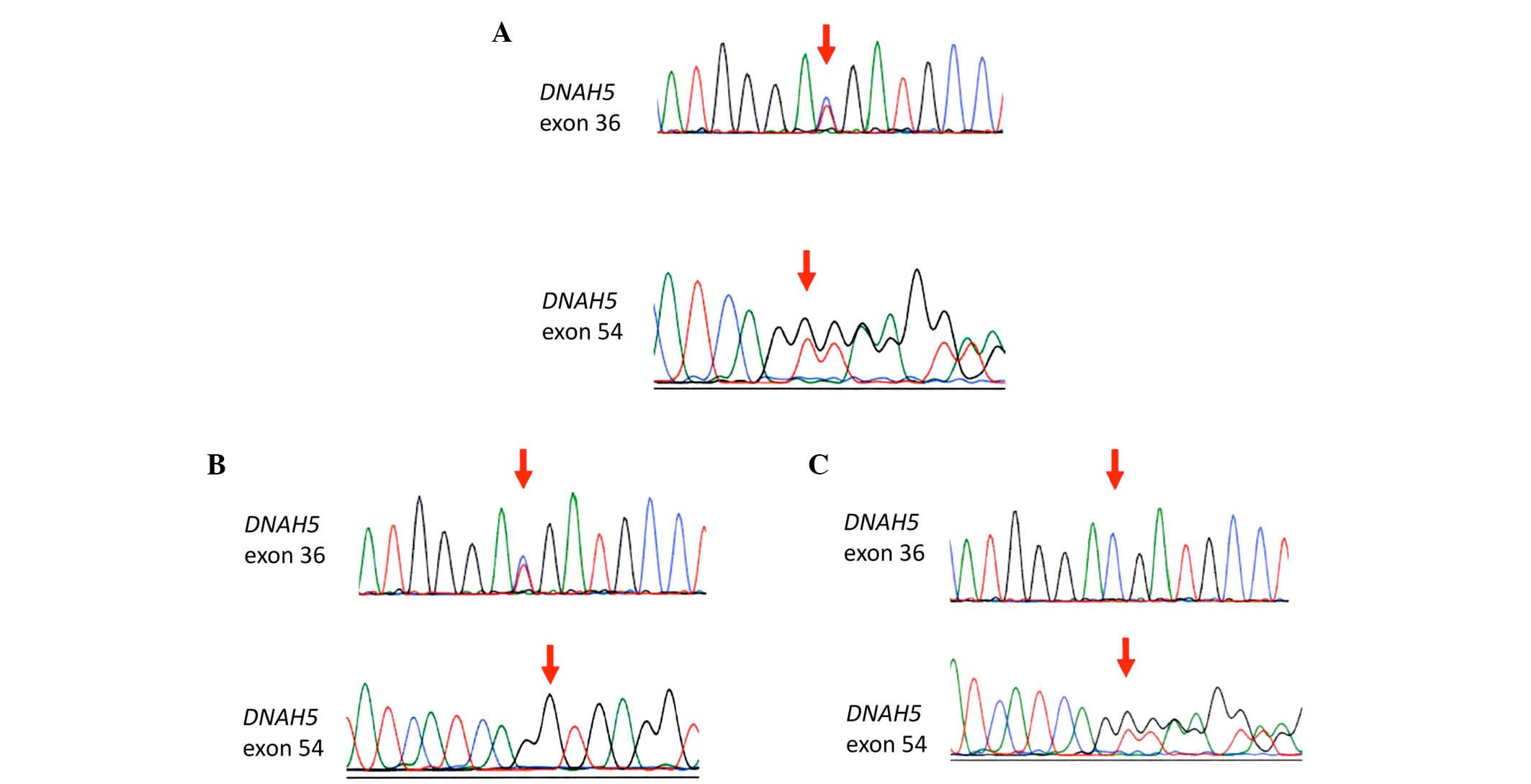
Sanger sequencing confirmed the compound
heterozygous mutations in DNAH5 identified by the
whole-exome analysis in the proband (Fig. 7A); NM_001369.2:c.5983C>T,
p.Arg1995X in exon 36 and NM_001369.2:c.9101delG, p.Gly3034ValfsX22
in exon 54. The patient's father carried only the former mutation
(Fig. 7B), and his mother carried
only the latter mutation (Fig.
7C). These observations confirmed that each mutation was
inherited from a different parent.
Discussion
Pediatricians had followed this case for nine years
before the diagnosis of PCD was made. Reportedly, the majority of
patients with PCD are seen by physicians greater than 50 times
prior to a diagnosis of PCD being made, and the mean age at PCD
diagnosis is 10.9±14.4 years (2).
In addition, it takes markedly longer to make a diagnosis of PCD
when situs inversus is absent. The value of this case is that
sequential chest X-rays and CT scans were obtained over a long time
period. An abnormality in a chest X-ray was observed at the age of
three, however not at the age of one, and at a later date the
patient had consolidation only in one lobe (the right middle lobe).
For subjects 5–11 years old, Davis et al (10) observed that patients with inner
dynein arm and central apparatus defects with microtubular
disorganization presented with more lobes with bronchiectasis
(median, 5; p=0.0008) and consolidation (median, 3; p=0.0001) than
patients with outer dynein arm defects (median, 3 and 2,
respectively). The proband in the current study had defects only in
the outer dynein arms; therefore, he had a less severe lung
pathology, which may partly explain the delay in a diagnosis of
PCD.
According to Santamaria et al (11), the prevalence of lung changes on CT
is as follows: Bronchiectasis, 80%; peribronchial thickening, 80%;
mucous plugging, 75%; parenchyma, 65%; and mosaic perfusion, 45%.
The lung CT of the patient in the present study indicated the
presence of bronchiectasis and peribronchial thickening, the two
most frequently occurring observations.
PCD is a Mendelian autosomal recessive and a
genetically heterogeneous disorder. In a review article from 2013,
Knowles et al (12)
reported that PCD-causing mutations had been identified in 21
genes. The genes most commonly identified to result in PCD were
DNAH5 (15–21%), DNAI1 (2–9%), DNAAF1 (LRRC50)
(4–5%), CCDC39 (2–10%), CCDC40 (2–8%), DNAH11
(6%) and LRRC6 (3%). Pathogenic mutations in 28 genes can
reportedly lead to PCD, and these 28 genes reportedly account for
approximately 70% of individuals affected with PCD (13). In the current study, electron
microscopy identified shortened outer dynein arms. Regarding the
association between ultrastructural phenotypes and genotypes,
mutation of DNAH5, DNAI1, DNAI2, DNAL1, CCDC114, TXNDC3 or
ARMC4, which code for the structural components of the outer
dynein arms, results in their loss.
In the present study, whole-exome analysis of
proband genomic DNA identified compound heterozygous mutations in
DNAH5. At present, DNAH5 is reportedly the gene most
often responsible for PCD; for example, mutations on both alleles
of DNAH5 were identified in 15% of a clinically
heterogeneous cohort of patients (14).
DNAH5 is a large gene comprising 79 exons and
one alternative first exon and it encodes a heavy chain of the the
outer dynein arm (8). A 1.5-kb
partial cDNA representing DNAH5 was identified by Omran
et al (15), and a
full-length, 14 kb DNAH5 transcript was characterized by
Olbrich et al (16).
Hornef et al (8) used haplotype analyses and/or
sequencing to screen 109 caucasian PCD families originating from
Europe and North America for the presence of DNAH5
mutations. They identified 33 novel and 2 known DNAH5
mutations. They observed clustering of mutations within five exons
(exons 34, 50, 63, 76 and 77); and these five exons harbored 27
(52%) of all 52 detected mutant alleles. Based on these
observations, direct sequencing was conducted in the current study
with these five exons to screen PCD-causing mutations. However, the
two mutations in the examined patient were in exons 36 and 54:
NM_001369.2:c.5983C>T, p.Arg1995X in exon 36 and
NM_001369.2:c.9101delG, p.Gly3034ValfsX22 in exon 54.
Although DNAH5 mutations have been reported
in PCD patients outside Japan, only one report of DNAH5
mutation in a Japanese patient was identified in PubMed. Tate et
al (17) analyzed the case of
a male neonate who exhibited three lobes of the left lung, asplenia
and complex heart anomalies, who died 6 h subsequent to delivery. A
heterozygous single nucleotide change (c.7829A>G) was identified
in exon 47 of DNAH5, and this mutation resulted in the
missense mutation of p.Glu2610Gly (17).
Zhang et al (18) performed exome capture and
sequencing with samples from one affected individual and the
unaffected parents from a Chinese Han community. They identified a
homozygous mutation, c. 8030G>A (Arg2677Gln), in DNAH5.
This mutation was in exon 49, which is a known hot spot for
PCD-causing mutations. Additionally, a patient from Germany had a
missense mutation in 8029C>T, which resulted in Arg2677X
(8).
To the best of our knowledge, both mutations
identified in the current study are novel (5,8,14,16).
One is a nonsense mutation (NM_001369.2:c.5983C>T, p.Arg1995X)
in exon 36; the other is a frame-shift mutation
(NM_001369.2:c.9101delG, p.Gly3034ValfsX22) in exon 54. Of the
PCD-causing mutations analyzed thus far, 85% are loss-of-function
variants, and approximately 15% are conservative missense mutations
(12). Among the 33 novel
DNAH5 mutations detected by Hornef et al (8), 12 were nonsense mutations; 8
frame-shift mutations; 5 splicing variants and 8 missense
mutations.
To predict the impact of these two newly identified
variants in silico, MutationTaster (19) and Polyphen-2 (20) analyses were conducted. PolyPhen-2
analysis indicated that the p.Gly3034ValfsX22 mutation of
DNAH5 was likely to be functionally damaging, with a score
of 1.000. MutationTaster assesses whether the predicted mutant
proteins will be long- or short-lived and whether nonsense-mediated
mRNA decay is likely to occur. MutationTaster predicted that each
of these DNAH5 mutations would cause nonsense-mediated mRNA
decay.
DNAH5 encodes ciliary dynein axonemal heavy
chain 5, a 4624-amino acid protein (16). The N-terminal domain forms the stem
domain of the outer dynein arm complex and is involved in
interactions with other heavy, intermediate and light chains. The
C-terminal region that constitutes the globular head contains six
conserved 6 p-loop domains and a conserved microtubule binding site
(16). The first p-loop domain is
known to bind and hydrolyze adenosine triphosphate (16). Both newly identified mutations were
predicted to cause loss of function of the protein; therefore, it
is likely that the two novel mutations are causal mutations
resulting in PCD.
This is, to the best of our knowledge, the first
report describing DNAH5 mutations in a Japanese patient with
PCD. Whether there are relatively fewer patients with PCD among the
Japanese compared with other ethnic groups is unclear, and requires
investigation in future studies.
The present study reported a boy who had been
followed by pediatricians for the first nine years of his life.
Electron microscopy identified loss of the outer dynein arms in the
cilia analyzed. Whole-exome analysis of the genomic DNA identified
novel compound heterozygous mutations in DNAH5:
NM_001369.2:c.5983C>T, p.Arg1995X in exon 36; and
NM_001369.2:c.9101delG, p.Gly3034ValfsX22 in exon 54.
Acknowledgements
The present study was supported by Grant-in-Aid for
General Scientific Research (C; grant nos. 25462662 and 16K11210)
from the Ministry of Education, Sciences and Culture of Japan and
the budget allocation from the director of Mie University Hospital
(2013, 2014). The authors would like to thank Dr Issei Kobayashi
and Dr Yuhko Kobayashi of Core-Lab, Graduate School of Regional
Innovation Studies, Mie University (Mie, Japan) for their
assistance with the genetic analysis. An English Language editing
service provided by Forte Science Communications (Tokyo, Japan) was
used (job no. R1506890).
References
|
1
|
Afzelius BA and Mossberg B: Immotile-cilia
syndrome (primary ciliary dyskinesia), including Kartagener
syndromeThe Metabolic and Molecular Bases of Inherited Disease.
Scriver C, Beaudet A, Sly W and Valle D: McGraw-Hill; New York: pp.
3943–3954. 1995
|
|
2
|
Sommer JU, Schäfer K, Omran H, Olbrich H,
Wallmeier J, Blum A, Hörmann K and Stuck BA: ENT manifestations in
patients with primary ciliary dyskinesia: Prevalence and
significance of otorhinolaryngologic co-morbidities. Eur Arch
Otorhinolaryngol. 268:383–388. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ellerman A and Bisgaard H: Longitudinal
study of lung function in a cohort of primary ciliary dyskinesia.
Eur Respir J. 10:2376–2379. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rubin BK: Immotile cilia syndrome (primary
ciliary dyskinesia) and inflammatory lung disease. Clin Chest Med.
9:657–668. 1988.PubMed/NCBI
|
|
5
|
Djakow J, Svobodová T, Hrach K, Uhlík J,
Cinek O and Pohunek P: Effectiveness of sequencing selected exons
of DNAH5 and DNAI1 in diagnosis of primary ciliary dyskinesia.
Pediatr Pulmonol. 47:864–875. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
American Thoracic Society; European
Respiratory Society, . ATS/ERS recommendations for standardized
procedures for the online and offline measurement of exhaled lower
respiratory nitric oxide and nasal nitric oxide, 2005. Am J Respir
Crit Care Med. 171:912–930. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Narang I, Ersu R, Wilson NM and Bush A:
Nitric oxide in chronic airway inflammation in children: Diagnostic
use and pathophysiological significance. Thorax. 57:586–589. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hornef N, Olbrich H, Horvath J, Zariwala
MA, Fliegauf M, Loges NT, Wildhaber J, Noone PG, Kennedy M,
Antonarakis SE, et al: DNAH5 mutations are a common cause of
primary ciliary dyskinesia with outer dynein arm defects. Am J
Respir Crit Care Med. 174:120–126. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zariwala MA, Leigh MW, Ceppa F, Kennedy
MP, Noone PG, Carson JL, Hazucha MJ, Lori A, Horvath J, Olbrich H,
et al: Mutations of DNAI1 in primary ciliary dyskinesia: Evidence
of founder effect in a common mutation. Am J Respir Crit Care Med.
174:858–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Davis SD, Ferkol TW, Rosenfeld M, Lee HS,
Dell SD, Sagel SD, Milla C, Zariwala MA, Pittman JE, Shapiro AJ, et
al: Clinical features of childhood primary ciliary dyskinesia by
genotype and ultrastructural phenotype. Am J Respir Crit Care Med.
191:316–324. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Santamaria F, Montella S, Tiddens HA,
Guidi G, Casotti V, Maglione M and de Jong PA: Structural and
functional lung disease in primary ciliary dyskinesia. Chest.
134:351–357. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Knowles MR, Daniels LA, Davis SD, Zariwala
MA and Leigh MW: Primary ciliary dyskinesia. Recent advances in
diagnostics, genetics, and characterization of clinical disease. Am
J Respir Crit Care Med. 188:913–922. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Knowles MR, Ostrowski LE, Leigh MW, Sears
PR, Davis SD, Wolf WE, Hazucha MJ, Carson JL, Olivier KN, Sagel SD,
et al: Mutations in RSPH1 cause primary ciliary dyskinesia with a
unique clinical and ciliary phenotype. Am J Respir Crit Care Med.
189:707–717. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Failly M, Bartoloni L, Letourneau A, Munoz
A, Falconnet E, Rossier C, de Santi MM, Santamaria F, Sacco O,
DeLozier-Blanchet CD, et al: Mutations in DNAH5 account for only
15% of a non-preselected cohort of patients with primary ciliary
dyskinesia. J Med Genet. 46:281–286. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Omran H, Häffner K, Völkel A, Kuehr J,
Ketelsen UP, Ross UH, Konietzko N, Wienker T, Brandis M and
Hildebrandt F: Homozygosity mapping of a gene locus for primary
ciliary dyskinesia on chromosome 5p and identification of the heavy
dynein chain DNAH5 as a candidate gene. Am J Respir Cell Mol Biol.
23:696–702. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Olbrich H, Häffner K, Kispert A, Völkel A,
Volz A, Sasmaz G, Reinhardt R, Hennig S, Lehrach H, Konietzko N, et
al: Mutations in DNAH5 cause primary ciliary dyskinesia and
randomization of left-right asymmetry. Nat Genet. 30:143–144. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tate G, Tajiri T, Kishimoto K and Mitsuya
T: A novel mutation of the axonemal dynein heavy chain gene 5
(DNAH5) in a Japanese neonate with asplenia syndrome. Med Mol
Morphol. 48:116–122. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang J, Guan L, Wen W, Lu Y, Zhu Q, Yuan
H, Chen Y, Wang H, Zhang J and Li H: A novel mutation of DNAH5 in
chronic rhinosinusitis and primary ciliary dyskinesia in a Chinese
family. Eur Arch Otorhinolaryngol. 271:1589–1594. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Adzhubei I, Schmidt S, Peshkin L, Ramensky
VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A method
and server for predicting damaging missense mutations. Nat Methods.
7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|