Introduction
The partial deletion of distal chromosome 10q was
first reported by Lewandowski et al (1), and the existence of a distinct
chromosome 10q26 deletion syndrome (Online Mendelian Inheritance in
Man no. 609625) was then suggested due to consistent clinical
findings (2–4). This syndrome is caused by a rare
chromosomal abnormality, although ≥110 cases have been reported in
the literature. Patients with this syndrome present with symptoms
that span a relatively extensive and heterogeneous phenotypic
spectrum, although common clinical features, including craniofacial
anomalies, developmental delay/intellectual disability (DD/ID),
urinary tract abnormalities, cardiac malformations and
neurodevelopmental deficits, have been observed (4–7). A
pure distal 10q deletion may be derived from familial or de
novo structural variation and feature breakpoints at 10q25 or
10q26, although interstitial or terminal deletions of large size,
which are typically identified through conventional cytogenetic
techniques, can also occur (4,8).
Chromosomal microarray analysis (CMA), including the use of array
comparative genome hybridization and single-nucleotide polymorphism
(SNP) arrays, has been recommended as the first-tier test for
individuals with DD/ID and/or congenital anomalies (9). CMA has been increasingly employed to
refine the location and size of 10q26 deletions to improve
understanding of genotype-phenotype correlations by identifying the
minimal critical region (CR) or the smallest region of deletion
overlap associated with 10q26 deletion syndrome. Because this
syndrome is a clinically heterogeneous disorder involving deletions
of variable locations and sizes, the CR for 10q26 deletion syndrome
and candidate genes for variable phenotypes remain unclear
(10). Therefore, further studies
on patients with 10q26 deletions should be performed to elucidate
the correlation between various phenotypes and the CR. The majority
of examined cases of this syndrome were previously characterized
using traditional techniques with limited resolution and low
efficiency, including G-banded karyotyping, fluorescence in
situ hybridization and microsatellite marker genotyping,
whereas few CMA-based molecular studies have been performed to
investigate these cases. The current study attempted to refine a CR
for this syndrome by performing a high-resolution molecular
analysis of two unrelated patients with pure terminal 10q26
deletions and comparing these patients with patients with pure
distal 10q deletions described in previous CMA-based studies and
the Database of Chromosomal Imbalance and Phenotype in Humans using
Ensembl Resources (DECIPHER; https://decipher.sanger.ac.uk/), specifically
including patients from this database with deleted regions
encompassed by the deleted regions of the two patients examined in
the current study and that presented without any other copy number
variants or with other likely benign copy number variants. In
addition, the phenotypic variability associated with CRs proposed
in previous studies is discussed.
Materials and methods
Case presentation
Patient 1, a 3-year-old female, was the second child
of healthy, unrelated parents. Prenatal ultrasonography indicated
that duodenal atresia was present in the fetus; therefore, the
karyotyping of fetal blood was performed. The fetal karyotype did
not reveal any chromosomal abnormalities (data not shown). Thus,
the patient's parents continued the pregnancy. Premature rupture of
membranes occurred at 36 weeks of gestation. Eventually, the
parents opted for delivery by caesarian section. No neonatal
respiratory distress was observed. The infant's weight and length
at birth were 2,420 g (<50th percentile) and 46 cm (<50th
percentile), respectively. Surgical therapy for duodenal atresia
was administered following birth, and an operation to repair patent
ductus arteriosus (PDA) was performed at 12 days after birth. The
child was referred to the Fetal Medicine Center (The First
Affiliated Hospital of Sun Yat-Sen University, Guangzhou) due to DD
and mild ID observed at 3 years and 9 months of age. Milestones
were significantly delayed, with walking occurring at 3 years;
current, the patient has no speech. The patient's psychomotor
development was significantly delayed. At the age of 3 years and 9
months, her weight, height and head circumference were 9.6 kg (3rd
percentile), 83.6 cm (3rd percentile) and 46.5 cm (<10th
percentile), respectively. Cerebral magnetic resonance imaging did
not reveal any abnormalities at this time. The patient exhibited
certain craniofacial features of 10q26 deletion syndrome, including
a triangular-shaped face, hypertelorism, strabismus, a broadly
prominent nose and micrognathia. Palate anomalies were not
observed. Her ear shape was normal, and audiometry indicated that
she possessed normal hearing. An image of the patient is not
available in the present report. Patient 2, a 5-year-old female,
was the second child of healthy, unrelated parents. She was born at
full term by caesarian section due to a scarred maternal uterus.
Her birth weight and length were 2,450 g (<3rd percentile) and
47 cm (<10th percentile), respectively. Congenital heart defects
(CHDs), including atrial septal defect and PDA, were observed by
ultrasound examination during the neonatal period. At 3 months, the
patient underwent an operation to address the CHDs. She walked and
spoke her first word at 3 years. At the age of 5 years and 2
months, her weight, height and head circumference were 13 kg (3rd
percentile), 96 cm (3rd percentile) and 47.6 cm (<10th
percentile), respectively. The patient displayed normal motor
skills and performed well at ice-skating at this time. However,
mild ID and hyperactive behavior were observed. Craniofacial
features of 10q26 deletion syndrome, including a high and narrow
palatine arch, a triangular-shaped face, low-set ears,
hypertelorism, strabismus, a broadly prominent nose and
micrognathia, were observed. The patient's hearing was normal.
Again, an image of the patient is not available in the present
report.
Cytogenetic analysis
Using standard procedures, routine G-banded
karyotyping was performed on peripheral blood specimens from each
child and her parents, as previously described (11).
High-resolution SNP array
analysis
Genomic DNA was isolated from peripheral blood
lymphocytes with the QIAamp DNA Blood Mini kit (Qiagen, Inc.,
Valencia, CA, USA) in accordance with the manufacturer's
instructions. The SNP array was performed according to the
manufacturer's protocols. Briefly, genomic DNA samples (250 ng)
were digested, amplified and labeled. The DNA was hybridized to
CytoScan HD array on the GeneChip Hybridization Oven 645
(Affymetrix, Inc., Santa Clara, CA, USA) at 50°C for 16–18 h,
washed on the GeneChip Fluidics Station 450 (Affymetrix, Inc.),
stained with Affymetrix GeneChip Stain Reagents, and scanned on the
GeneChip Scanner 3000 7G (Affymetrix, Inc.). The raw data were
analyzed using Chromosome Analysis Suite 3.0 software (ChAS 3.0;
Affymetrix, Inc.) The CytoScan HD array contains more than 2.6
million markers for copy number analysis; 1,950,000 of these
markers are unique, non-polymorphic oligonucleotide probes, and
750,000 markers are SNP probes used for genotyping. The average
marker spacing is one probe every 1.1 kb, with a mean spacing of
one probe every 1.7 kb on non-gene backbones and one probe every
880 bp in intragenic regions. Using ChAS 3.0 software (Affymetrix,
Inc.), aberrations were filtered up to a minimal size of 100 kb,
with ≥50 probe calls for deletions and duplications.
RefSeq and Database of Chromosome
Imbalance and Phenotype in Humans Using Ensemble Resources
(DECIPHER)
The building of the human genome assembly was based
on Homo sapiens GRCH37/hg19. The gene content of the deleted
region, including Online Mendelian Inheritance in Man genes and
RefSeq genes, was viewed and assessed with the UCSC Genome Browser
(genome.ucsc.edu). The location and size of the
deleted region was compared with similar cases described in
previous CMA-based studies and the DECIPHER (decipher.sanger.ac.uk/).
Results
Routine karyotyping of the two patients revealed
similar karyotypes of 46, XX, del(10) (q26) (Fig. 1A). The parents of the patients
presented with normal chromosome karyotypes, demonstrating the
de novo origin of the examined 10q26 deletions.
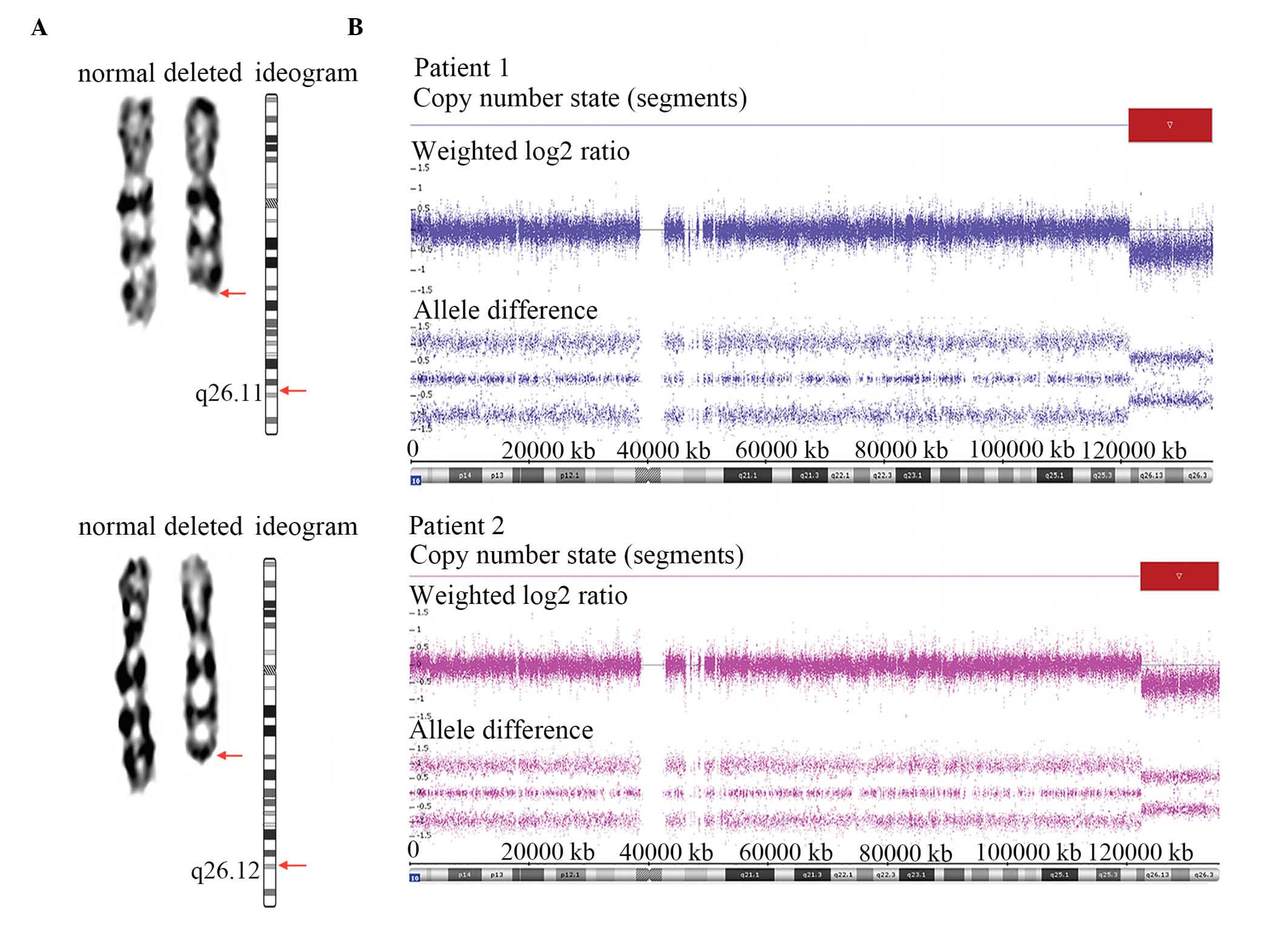 | Figure 1.Results of karyotyping and SNP array
analysis in Patient 1 and Patient 2. (A) Karyograms and ideograms
of chromosome 10 in Patient 1 and Patient 2. Arrows indicate the
locations of breakpoints. The karyotypes of both patients were 46,
XX, del (10)(q26). (B) The SNP
array defined a deleted genomic region from 10q26 to 10qter in the
two examined patients. The findings for Patients 1 and 2 were
analyzed using ChAS 3.0 software. Both log2 ratios and SNP
genotyping calls accurately indicate the locations and sizes of the
deleted regions. The red box outlines the deleted regions. SNP
array analysis refined the results, specifically indicating that
the deleted region of Patient 1 was del(10)(q26.11), which spans a ~14.04 Mb
segment (chr10:121,385,398–135,427,143), and that the deleted
region of Patient 2 was del (10)(q26.12), which spans a ~13.04 Mb
segment (chr10:122,387,570–135,427,143). SNP, single nucleaotide
polymorphism. |
In the SNP array for Patient 1, a 10q terminal
deletion with a breakpoint at 10q26.11 was detected (Fig. 1B). The deleted region was ~14.04 Mb
in size (chr10: 121,385,398–135,427,143) and contained ~117 RefSeq
genes (~83 known coding genes). In the SNP array for Patient 2, the
10q terminal deletion was ~13.04 Mb in size (chr10:
122,387,570–135,427,143) with a breakpoint at 10q26.12 (Fig. 1B). The deleted region encompassed
~108 RefSeq genes (~78 known coding genes). Patient 1 exhibited a
larger proximally deleted region compared with Patient 2. Thus, the
location of the deletion (chr10: 121,385,398–135,427,143) in
Patient 1 was used as a search index in the DECIPHER database. The
eventual results of this search indicated that the deleted regions
of 30 previous cases involving 10q26 deletion were encompassed by
this region (Fig. 2). These cases
shared certain phenotypes with 10q26 deletion syndrome.
Furthermore, to the best of our knowledge, ≥18 cases of 10q26
deletions have been characterized by CMA in prior studies (Table I). The clinical features and
locations of the 10q26 deletions of patients from the current
study, previous studies and the DECIPHER database are reviewed in
Fig. 2 and Table I in an attempt to map or refine a
CR.
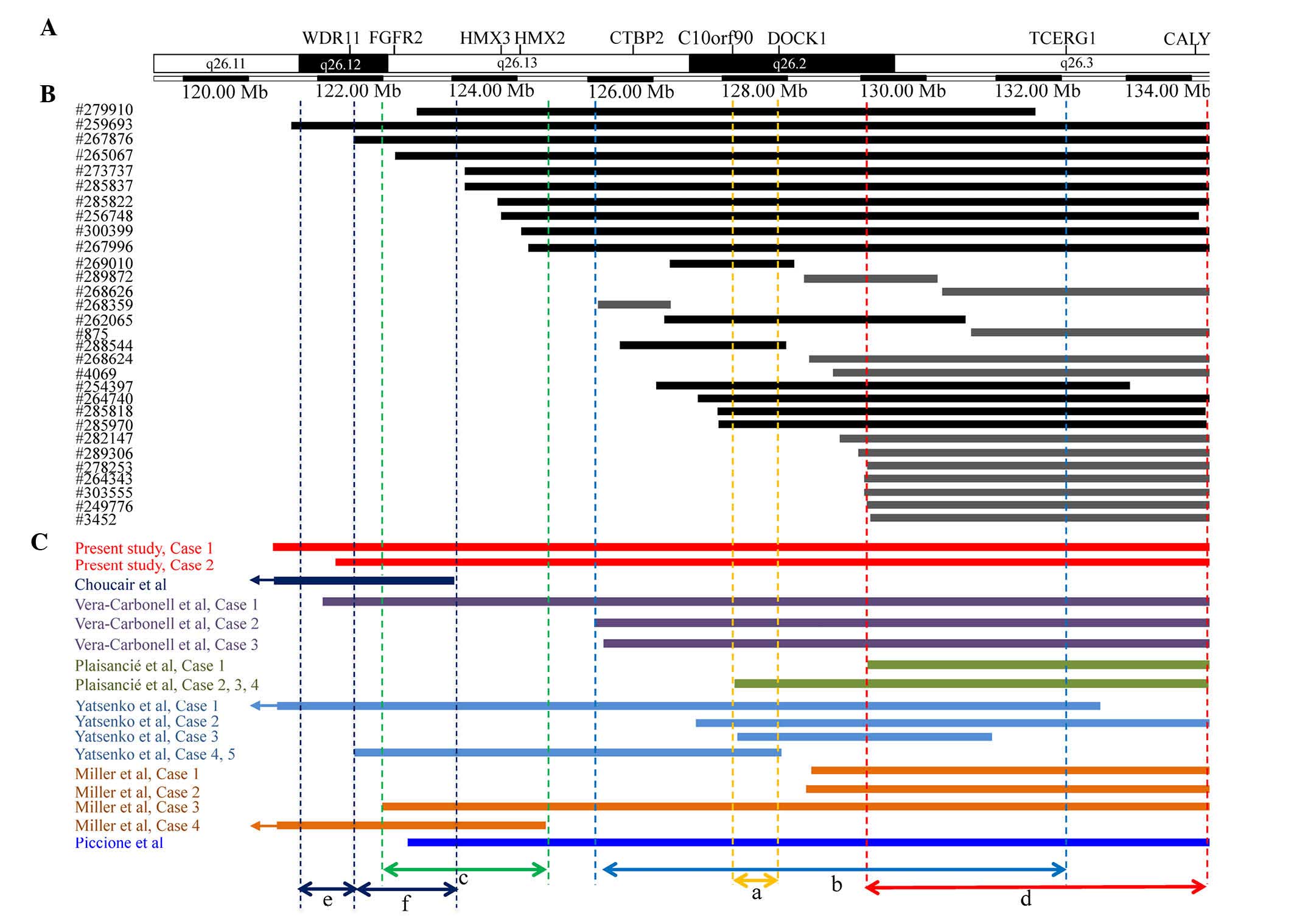 | Figure 2.An overlapping map of 10q26 deletions
in CMA-assessed patients described in the present study, prior
studies and the DECIPHER database. (A) A schematic presentation of
the 10q26 region with gene positions as specified by the University
of California Santa Cruz Genome Browser. Double-headed arrows
between dotted lines indicate different CRs; a=the CR proposed by
Yatsenko et al (7) for
common clinical features of 10q26 deletion syndrome; b=the CR
proposed by Vera-Carbonell et al (10) for renal/urinary anomalies; c=the CR
proposed by Miller et al (12) for inner ear malformation,
vestibular dysfunction and hearing loss; d=the deleted region
suggested by the current study as a region that may contribute to
common clinical features of 10q26 deletion syndrome; e=the CR
proposed by Choucair et al (13) for genital anomalies; and f=the CR
proposed by Choucair et al (13) for common clinical features. (B) An
overview of patients from the DECIPHER database with distal 10q26
deletions. Both black and gray vertical bars represent deleted
regions; the black bars overlap with the CR proposed by Yatsenko
et al (7), but the gray
bars do not. Of 13 patients represented by gray vertical bars, 12
patients overlap with the deletion with a breakpoint at
approximately 130.0 Mb instead of the CR proposed by Yatsenko et
al (7). The majority of these
12 patients presented with common features of 10q26 deletion
syndrome. (C) An overview of CMA-assessed patients with 10q26
deletions described in various reports. Only four cases, including
cases 1, 2 and 4 described by Miller et al (12) and case 1 described by Plaisancié
et al (14), exhibited
common clinical features of 10q26 deletion syndrome and harbored
the smallest terminal deletion with a breakpoint at approximately
130.0 Mb. CMA, chromosomal microarray analysis; CR, critical
region; FGFR2, fibroblast growth factor receptor 2; CTBP2,
C-terminal binding protein 2; DOCK1, dedicator of cytokinesis 1;
TCERG1, transcription elongation regulator 1; CALY, calcyon neuron
specific vesicular protein. |
 | Table I.Clinical features in patients with
10q26 deletions characterized by chromosomal microarray analysis in
the present study and previous studies. |
Table I.
Clinical features in patients with
10q26 deletions characterized by chromosomal microarray analysis in
the present study and previous studies.
|
|
|
|
|
|
|
| Present study |
|---|
| Feature | Yatsenko et
al, 2009 (7) | Vera-Carbonell et
al, 2015 (10) | Miller et al,
2009 (12) | Choucair et
al, 2015 (13) | Plaisancié et
al, 2014 (14) | Piccione et
al, 2008 (15) | Patient 1 | Patient 2 |
|---|
| Total number of
patients | 5 | 3 | 4 | 1 | 4 | 1 |
|
|
| Gender | 3 M, 2 F | 0 M, 3 F | 2 M, 2 F | 1 M | 0 M, 4 F | 1 M, 0 F | 1 F | 1 F |
| Deletion size
(Mb) | 3.51–17.22 | 9.17–13.46 | 6.08–12.45 | 4.5 | 4.96, 7.09 | 11.4 | 14.04 | 13.04 |
| Craniofacial
dysmorphism | 5/5 | 3/3 | 4/4 | 1/1 | 4/4 | 1/1 | + | + |
|
Microcephaly | 4/5 | 2/3 |
| 1 | 3/4 | 1/1 |
|
|
|
Brachycephaly |
|
| 1/4 |
|
|
|
|
|
|
Craniosynostosis |
|
| 1/4 |
|
|
|
|
|
| Widow's
peak |
|
| 1/4 |
|
|
|
|
|
| Tall
forehead |
|
| 3/4 |
| 4/4 |
|
Bitemporal narrowing |
|
|
|
| 3/4 |
|
|
|
| Ear
anomalies | 4/5 | 3/3 | 2/4 | 1/1 | 4/4 | 1/1 | + | + |
| Thick
eyebrows |
|
|
|
| 1/4 |
|
|
|
| Deep
set eyes |
|
|
|
| 2/4 |
|
|
|
|
Strabismus | 2/5 | 3/3 | 4/4 | 1/1 |
| 1/1 | + | + |
|
Broad/prominent nose | 5/5 | 3/3 | 2/4 | 1/1 | 4/4 | 1/1 | + | + |
| Choanal
atresia |
|
|
|
|
| 1/1 |
|
|
|
Hypertelorism | 2/3 |
| 1/4 |
| 1/4 | 1/1 | + | + |
| Long
philtrum | 1/5 | 2/3 | 1/4 |
| 4/4 |
| + | + |
| Thin
vermilion of the lips | 2/5 | 2/3 | 1/4 | 1/1 | 4/4 |
|
|
|
|
Downturned corners of the
mouth | 1/5 |
|
|
|
|
|
|
|
| Bifid
uvula |
|
|
|
| 1/4 |
|
|
|
|
Posterior cleft palate |
|
|
|
| 1/4 |
|
|
|
| High
arched palate |
|
| 1/4 |
|
|
| + |
|
| Broad
chin |
| 1/3 |
|
| 4/4 |
|
|
|
|
Micrognathia |
|
| 1/4 |
|
| 1/1 | + | + |
| Neurologic
abnormalities | 4/5 | 3/3 | 4/4 | 1/1 | 4/4 | 1/1 | + | + |
|
Developmental delay | 4/5 | 3/3 | 4/4 | 1/1 | 4/4 | 1/1 | + | + |
|
Intellectual disability | 3/5 | 3/3 | 3/4 | 1/1 | 4/4 |
| + | + |
|
Behavioral problems | 3/5 |
|
|
| 4/4 |
|
| + |
|
Seizure |
|
|
|
|
| 1/1 |
|
|
|
Hypotonia |
| 3/3 | 3/4 | 1/1 |
| 1/1 |
|
|
| Limb anomalies | 2/5 | 2/3 | 4/4 |
| 3/4 |
|
|
|
|
Clinodactyly | 2/5 | 2/3 | 2/4 |
| 3/4 |
|
|
|
|
Tapering of the fingers | 1/5 |
| 1/4 |
|
|
|
|
|
| Short
fifth metacarpal bones | 1/5 |
|
|
| 1/4 |
|
|
|
|
Brachydactyly |
|
|
|
| 1/4 |
|
|
|
|
Planovalgus foot |
|
| 1/4 |
| 1/4 |
|
|
|
|
Overlapping toes |
|
| 1/4 |
|
|
|
|
|
|
Hypoplastic toe nails |
|
| 1/4 |
|
|
|
|
|
| Congenital heart
disease | 3/5 |
| 1/4 |
|
| 1/1 | + | + |
| Patent
ductus arteriosus | 3/5 |
|
|
|
|
| + | + |
|
Ventricular septal defect |
|
| 1/4 |
|
| 1/1 |
|
|
| Atrial
septal defect |
|
|
|
|
|
|
| + |
|
Ventricular asymmetry |
|
|
|
| 1/4 |
|
|
|
|
| Urinary tract/renal
anomalies | 2/5 | 2/3 | 1/4 |
|
|
|
|
|
|
Hydronephrosis | 1/5 |
| 1/4 |
|
|
|
|
|
| Dilated
right ureter | 1/5 |
|
|
|
|
|
|
|
| Right
kidney | 1/5 |
|
|
|
|
|
|
|
| Small
kidneys |
| 1/3 |
|
|
|
|
|
|
|
Vesicoureteral reflux |
| 1/3 |
|
|
|
|
|
|
| Genital
anomalies | 1/5 | 2/3 | 2/4 | 1/1 |
| 1/1 |
|
|
|
Cryptorchidism | 1/5 |
|
| 1/1 |
| 1/1 |
|
|
|
Ambiguous genitalia/sex
reversal | 1/5 |
| 1/4 |
|
|
|
|
|
| Right
undescended testicle |
|
| 1/4 |
|
|
|
|
|
| Small
labia minora and majora |
| 1/3 |
|
|
|
|
|
|
| Small
labia majora |
| 1/3 |
|
|
|
|
|
|
| CNS
malformations | 1/5 | 1/3 |
|
| 1/4 |
|
|
|
|
| Global
cerebellar hypoplasia |
|
|
|
| 1/4 |
|
|
|
| Pineal
cyst | 1/5 |
|
|
|
|
|
|
|
|
Enlargement of the cisterna
magna |
| 1/3 |
|
|
|
|
|
|
| Hearing loss | 1/5 | 1/3 | 2/4 |
|
|
|
|
|
| Duodenal
atresia |
|
|
|
|
|
| + |
|
Discussion
In the present study, the deleted regions from two
patients with 10q26 terminal deletions were molecularly
characterized using a high-resolution SNP array. This approach
allowed for the accurate determination of the locations and sizes
of these regions. The two patients shared various clinical features
associated with 10q26 deletion syndrome, including DD/ID, growth
retardation, craniofacial dysmorphism and CHDs. The findings from
these patients also emphasized several of the common clinical
characteristics observed in patients with pure 10q26 deletions.
The severity and extent of clinical characteristics
associated with 10q26 deletions may depend on the varying locations
and sizes of these deletions and the number of haploinsufficient
genes in deleted regions. The characterization of atypical,
overlapping and distinct deletions may lead to a map of CRs or even
the identification of candidate genes in particular cases. Although
correlations between the location and size of 10q26 deletions and
phenotypic variations remain incompletely elucidated, ≥five CRs in
the 10q26 region have been previously hypothesized for 10q26
deletion syndrome (7,10,12,13).
Yatsenko et al (7) proposed
a ~600 kb CR (Fig. 2A) at 10q26.2
for common clinical features, including craniofacial dysmorphism,
CHD and DD/ID. This proposed CR contains two protein-coding genes:
The chromosome 10 open reading frame 90 (C10orf90) gene and
the dedicator of cytokinesis 1 (DOCK1) gene. The latter gene
influences various cellular biological processes, including
phagocytosis, cell migration, apoptosis and tumorigenesis, among
others. The DOCK1 gene is also thought to be important in
the pathogenesis of cardiovascular and urinary anomalies. Thus,
Yatsenko et al (7)
suggested the DOCK1 gene as a candidate gene for 10q26
deletion syndrome. The findings of the current study have further
supported the idea that this proposed CR (Fig. 2A) may be important in 10q26
deletion syndrome, as the majority of the clinical features of the
patients assessed in the present study were consistent with the
presence of smaller deleted regions that overlap with this CR.
Notably, the current study identified that 5 patients described in
previous studies [cases 1, 2 and 4 reported by Miller et al
(12) and case 1 reported by
Plaisancié et al (14) and
a case described by Choucair et al (13)] and 13 patients (Fig. 2B; represented by gray vertical
bars) in the DECIPHER database did not have deletions that
overlapped with this CR (Fig. 2A),
although the majority of these patients presented with common
features of 10q26 deletion syndrome. Based on the overlapping map
generated in this study (Fig. 2),
it was demonstrated that 15 patients, including 3 patients reported
in previous studies [cases 1 and case 2 reported by Miller et
al (12) and case 1 reported
by Plaisancié et al (14)]
and 12 patients (Fig. 2B;
represented by gray vertical bars) in the DECIPHER database,
overlapped with a small terminal deletion with a breakpoint at ~130
Mb. However, the majority of these 15 patients presented with
common features of 10q26 deletion syndrome. Therefore, it is
reasonable to suspect that distal 10q26 harbors other CRs. We
hypothesize that the distal 10q26 terminal deletion with a
breakpoint at ~130.0 Mb may contribute to the common clinical
features of 10q26 deletion syndrome.
In addition to a CR for common characteristics of
this syndrome, other CRs for specific phenotypes are increasingly
being identified. Miller et al (12) described a common 2.5 Mb deletion
region (Fig. 2C) associated with
inner ear malformations, vestibular dysfunction and hearing loss in
two patients. However, 7 out of 10 patients from the current and
previous studies that had distal 10q26 deletions overlapping this
common 2.5 Mb region did not exhibit hearing impairment. These 7
patients included a patient described by Piccione et al
(15), patients 4 and 5 in a
report by Yatsenko et al (7), patient 1 in a report by
Vera-Carbonell et al (10),
a patient described by Choucair et al (13) and the two patients described in the
present report. A similar trend is observed for all cases recorded
in the DECIPHER database that involve a deletion in this region.
Notably, among three patients described by Vera-Carbonell et
al (10), patient 3 did not
harbor a deletion that overlapped with this region, but did exhibit
hearing loss, patient 2 did not harbor a deletion that overlapped
with this region or present with hearing loss, and patient 1
exhibited normal hearing, in spite of harboring a deletion in this
region. In the previous study, Vera-Carbonell et al
(10) focused on renal/urinary
tract anomalies associated with 10q26 deletions and suggested that
small 10q26.2 distal deletions may be associated with common
phenotypic characteristics rather than renal/urinary tract
anomalies, which are instead correlated with longer 10q26.2 distal
deletions (Fig. 2B). Notably,
among individuals with a 10q26 deletion overlapping the proposed CR
for renal/urinary tract anomalies (Fig. 2B), 13 out of 18 patients [not
including patient 4 in a report by Miller et al (12) and a case reported by Choucair et
al (13)] examined in the
present study and previous studies, and 24 out of 30 cases recorded
in the DECIPHER database, did not exhibit such anomalies. Recently,
Choucair et al (13) also
proposed a novel CR for genital abnormalities (Fig. 2E) and another different CR for
common clinical features (Fig.
2F). Through a review of the present study and previous
studies, it was observed that genital abnormalities were presented
in 4 out of 6 cases with a 10q26 deletion overlapping the above
proposed CR for genital abnormalities (Fig. 2E). However, genital abnormalities
were not observed in the two cases of the current study. In
addition to potential CRs, studies of 10q26 deletions have also
proposed candidate genes, including the DOCK1, fibroblast
growth factor receptor 2 (FGFR2), C-terminal binding protein
2 (CTBP2), calcyon neuron specific vesicular protein
(CALY), WD repeat domain 11 (WDR11), H6 family
homeobox 2 (HMX2) and HMX3 genes within these regions
(7,10,12–14).
Phenotypic effects correlated with CRs may primarily depend on the
number of haploinsufficient genes in deleted regions. However, to
the best of our knowledge, there is little available evidence to
confirm the pathogenicity of haploinsufficiency for either
candidate genes or other genes in the 10q26 region. Further
investigations should be performed to identify dosage-sensitive
genes in this region and thereby obtain an improved understanding
of the genotype-phenotype correlations associated with 10q26
deletions.
The conflicting findings discussed above may be
attributed to incomplete penetrance/variable expressivity or the
effects of other potential CRs. Furthermore, if potential CRs were
assumed to be independent genomic sites, a ‘two-hit’ model would
account for these discordances. This model would propose that the
combined effects of two or more deletions would result in
phenotypic variability (16,17).
Each CR may contribute to particular phenotypes in a correlative
manner; as a result, manifestations of the clinical characteristics
of 10q26 deletion syndrome would be expected to be variable or
incomplete in the patients described in these studies.
In conclusion, the current study reported two new
patients with 10q26 deletion syndrome and reviewed the literature
regarding this syndrome. Although an overlap map for CMA-assessed
cases from the present study, previous studies and the DECIPHER
database, was generated CR refinement could not be accomplished due
to the limited number of available cases and the varying and
atypical deletions observed in these cases. However, in addition to
the CR suggested by Yatsenko et al (7), the results of the current study
suggest that the distal 10q26 terminal deletion with a breakpoint
at ~130.0 Mb may also contribute to common clinical features of
10q26 deletion syndrome. In addition, the association between three
previously reported CRs and phenotypic variability were discussed
in detail, thus, facilitating improved comprehension of the
phenotypic heterogeneity of 10q26 deletion syndrome.
References
|
1
|
Lewandowski RC Jr, Kukolich MK, Sears JW
and Mankinen CB: Partial deletion 10q. Hum Genet. 42:339–343. 1978.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wulfsberg EA, Weaver RP, Cunniff CM, Jones
MC and Jones KL: Chromosome 10qter deletion syndrome: A review and
report of three new cases. Am J Med Genet. 32:364–367. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schrander-Stumpel C, Fryns JP and Hamers
G: The partial monosomy 10q syndrome: Report on two patients and
review of the developmental data. J Ment Defic Res. 35:259–267.
1991.PubMed/NCBI
|
|
4
|
Irving M, Hanson H, Turnpenny P, Brewer C,
Ogilvie CM, Davies A and Berg J: Deletion of the distal long arm of
chromosome 10; is there a characteristic phenotype? A report of 15
de novo and familial cases. Am J Med Genet A 123A. 153–163. 2003.
View Article : Google Scholar
|
|
5
|
Courtens W, Wuyts W, Rooms L, Pera SB and
Wauters J: A subterminal deletion of the long arm of chromosome 10:
A clinical report and review. Am J Med Genet A. 140:402–409. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Scigliano S, Gregoire MJ, Schmitt M,
Jonveaux PH and LeHeup B: Terminal deletion of the long arm of
chromosome 10. Clin Genet. 65:294–298. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yatsenko SA, Kruer MC, Bader PI, Corzo D,
Schuette J, Keegan CE, Nowakowska B, Peacock S, Cai WW, Peiffer DA,
et al: Identification of critical regions for clinical features of
distal 10q deletion syndrome. Clin Genet. 76:54–62. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kehrer-Sawatzki H, Daumiller E,
Müller-Navia J, Kendziorra H, Rossier E, du Bois G and Barbi G:
Interstitial deletion del(10)(q25.2q25.3 approximately 26.11)-case
report and review of the literature. Prenat Diagn. 25:954–959.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miller DT, Adam MP, Aradhya S, Biesecker
LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE,
Epstein CJ, et al: Consensus statement: Chromosomal microarray is a
first-tier clinical diagnostic test for individuals with
developmental disabilities or congenital anomalies. Am J Hum Genet.
86:749–764. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vera-Carbonell A, López-Gonzalez V,
Bafalliu JA, Ballesta-Martínez MJ, Fernández A, Guillén-Navarro E
and López-Expósito I: Clinical Comparison of 10q26 overlapping
deletions: Delineating the critical region for urogenital
anomalies. Am J Med Genet A 167A. 786–790. 2015. View Article : Google Scholar
|
|
11
|
Claussen U, Michel S, Mühlig P, Westermann
M, Grummt UW, Kromeyer-Hauschild K and Liehr T: Demystifying
chromosome preparation and the implications for the concept of
chromosome condensation during mitosis. Cytogenet Genome Res.
98:136–146. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Miller ND, Nance MA, Wohler ES,
Hoover-Fong JE, Lisi E, Thomas GH and Pevsner J: Molecular (SNP)
analyses of overlapping hemizygous deletions of 10q25.3 to 10qter
in four patients: Evidence for HMX2 and HMX3 as candidate genes in
hearing and vestibular function. Am J Med Genet A 149A. 669–680.
2009. View Article : Google Scholar
|
|
13
|
Choucair N, Ghoch J Abou, Fawaz A,
Mégarbané A and Chouery E: 10q26.1 Microdeletion: Redefining the
critical regions for microcephaly and genital anomalies. Am J Med
Genet A 167A. 2707–2713. 2015. View Article : Google Scholar
|
|
14
|
Plaisancié J, Bouneau L, Cances C, Garnier
C, Benesteau J, Leonard S, Bourrouillou G, Calvas P, Vigouroux A,
Julia S and Bieth E: Distal 10q monosomy: New evidence for a
neurobehavioral condition? Eur J Med Genet. 57:47–53. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Piccione M, Antona V, Piro E, Cavani S,
Malacarne M, Pierluigi M and Corsello G: 10qter deletion: A new
case. Am J Med Genet A 146A. 2435–2438. 2008. View Article : Google Scholar
|
|
16
|
Girirajan S, Rosenfeld JA, Coe BP, Parikh
S, Friedman N, Goldstein A, Filipink RA, McConnell JS, Angle B,
Meschino WS, et al: Phenotypic heterogeneity of genomic disorders
and rare copy-number variants. N Engl J Med. 367:1321–1331. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Girirajan S, Rosenfeld JA, Cooper GM,
Antonacci F, Siswara P, Itsara A, Vives L, Walsh T, McCarthy SE,
Baker C, et al: A recurrent 16p12.1 microdeletion supports a
two-hit model for severe developmental delay. Nat Genet.
42:203–209. 2010. View
Article : Google Scholar : PubMed/NCBI
|














