Introduction
Hypoxia frequently occurs in a wide range of solid
tumors. Hypoxia is responsible for the promotion of tumor motility,
invasion and metastasis, genomic instability and cell survival
responses, through the regulation of gene expression downstream of
hypoxia-inducible factor 1 (HIF1) (1–3).
HIF1 is a heterodimeric transcription factor composed of an ‘α’ and
a ‘β’ subunit. HIF1α is a cytoplasmic protein responsive to
O2 levels, whereas HIF1β is a nuclear protein, which is
constitutively expressed independent of O2 levels. Under
hypoxic conditions, HIF1α is translocated to the nucleus where it
forms a heterodimer with HIF1β subunits, resulting in the
activation of HIF1 and its complex downstream genetic program,
which may be responsible for numerous hypoxia-associated cellular
changes (4). Hypoxia has been
identified to promote metastasis of tumor cells by induction of
epithelial-mesenchymal transition (EMT) (5,6). EMT
is a process by which epithelial cells lose their cell polarity and
cell-cell adhesion, and gain migratory and invasive properties,
thus becoming mesenchymal stem cells. Therefore, EMT is an
essential process that results in tumor metastasis. At the
molecular level, EMT is characterized by decreases in the
expression levels of epithelial markers, such as E-cadherin, and an
increase in mesenchymal markers, such as vimentin and snail family
transcriptional repressor 1 (Snail1), as well as an increase in
migratory and invasive behavior (7–9).
Under hypoxic conditions, expression of the EMT marker proteins has
previously been observed to be mutually dependent on the expression
and activation of HIF1α (8,9).
Previous studies have demonstrated that hypoxia
stimulates the production of reactive oxygen species (ROS) in tumor
cells and hypoxia-generated intracellular ROS may activate and
stabilize HIF1α, thus contributing to tumor growth and metastasis,
in addition to tumor recurrence and therapeutic resistance
(10–13). In this regard, ROS-induced HIF1α
activation may be a major target for future cancer therapeutic
approaches.
The present study examined the effects of
hypoxia-induced intracellular ROS on the EMT of HT29 human
colorectal cancer cells in the presence or absence of the
antioxidant dieckol. The hypoxia-inducing agent, CoCl2
was used to induce an increase in ROS levels and HIF1α expression
in HT29 cells, and led to the transition of cellular phenotypes,
corresponding to the fundamental events in EMT. These effects were
markedly attenuated following dieckol treatment. Therefore, the
present study demonstrated that ROS-induced activation of HIF1α was
a key factor contributing to the induction of EMT in HT29
cells.
Materials and methods
Cell culture and reagents
HT29 human colorectal cancer cells, obtained from
the American Type Culture Collection (Manassas, VA, USA), were
cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), supplemented with 10%
heat-inactivated fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin and 100 mg/ml streptomycin,
in a 5% CO2 atmosphere at 37°C. For the cell-culture experiments,
cells were passaged three times and detached using Trypsin-EDTA.
Transwell migration and Matrigel invasion chambers were purchased
from BD Biosciences (Franklin Lakes, NJ, USA). Primary antibodies
against HIF1α (cat. no. 610958), E-cadherin (cat. no. 7870),
vimentin (cat. no. 5741), Snail1 (cat. no. AV33314) and β-actin
(cat. no. MAB1501) were obtained from BD Biosciences, Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA), Cell Signaling Technology,
Inc. (Danvers, MA, USA), and Sigma-Aldrich; Merck Millipore
(Darmstadt, Germany), respectively. Dihydroethidium (DHE) was
obtained from Molecular Probes (Thermo Fisher Scientific, Inc.).
Dieckol was prepared as previously described by Lee et al
(14). All remaining chemicals and
reagents were purchased form Sigma-Aldrich; Merck Millipore, unless
stated otherwise.
Cell viability assay
The cytotoxicity of CoCl2 and/or dieckol
was determined using the 3-(4,
5-dimetylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
formazan assay. HT29 cells were seeded in 12-well plates at a
density of 2.0×105 cells/well in DMEM containing 10%
FBS. The medium was changed 36 h after seeding to serum-free low
glucose (1,000 mg/l) DMEM and the cells were incubated with 0–200
µM CoCl2 for 36 h. The cells were then incubated with or
without 0–100 mg/ml (0–216 µM) dieckol for 12 h. Subsequently, the
medium was carefully removed and 160 µl MTT (0.5 mg/ml final
concentration) solution was added to each well prior to incubation
for an additional 4 h at 37°C in 5% CO2. The medium was aspirated
without the formazan crystals and 1 ml dimethyl sulfoxide was added
to each well. The absorbance was then measured using a microplate
reader (iMark; Bio-Rad Laboratories, Inc., Hercules, CA, USA) at
540 nm.
Cell migration and invasion assay
Cell migration was determined using a Transwell
chamber. HT29 cells were treated with or without 50 µM
CoCl2 in serum-free low glucose (1,000 mg/l) DMEM for 24
h. The cells were trypsinized and collected. Subsequently, 300 µl
cell suspension (1.5×105 cells) was incubated at 37°C in
serum-starved DMEM containing 50 µM CoCl2 and/or 25
mg/ml dieckol and added to the upper compartment of the Transwell
chamber in a humidified atmosphere with 5% CO2 for 24 h. DMEM
containing 10% FBS was used as a chemoattractant in the lower
compartment. Following a 24 h incubation at 37°C, cells on the
upper surface of the membrane were removed and the cells that had
migrated below the surface of the membrane were fixed with 3.7%
formaldehyde in phosphate-buffered saline (PBS), stained with
crystal violet (1 g dissolved in 10% ethanol) for 30 min, washed
twice with PBS and counted under a phase contrast microscope using
a 10x objective lens. The number of migrated cells in five randomly
selected fields were counted. The migration assays were performed
in triplicate.
For the invasion assay, cells were seeded in the
Transwell invasion chambers, which were coated with Matrigel (1
mg/ml). DMEM supplemented with 10% FBS was added to the lower
compartment of the chamber. Cells were serum-starved and treated
with 50 µM CoCl2 and/or 25 mg/ml dieckol as
aforementioned, then trypsinized and collected. Subsequently, 300
µl of each cell suspension was added to the upper compartment of
the chamber and incubated at 37°C in a humidified atmosphere with
5% CO2 for 24 h. Cells that had invaded below the
surface of the membrane were fixed, stained, washed, and counted.
The number of cells in five randomly selected fields was counted.
The invasion assays were performed in triplicate.
ROS quantification
HT29 cells (2.0×105 cells/well) were
serum-starved for 24 h and were then incubated in the presence or
absence of 50 µM CoCl2 for 36 h, with the medium being
replaced twice, followed by treatment with or without 25 mg/ml
dieckol for 3 h. Cellular ROS levels were determined using DHE
staining and the Muse oxidative stress kit (Merck Millipore)
according to the manufacturer's protocol.
Western blotting
Following the aforementioned treatments, cells were
washed twice with PBS, harvested and solubilized in 2X sodium
dodecyl sulfate (SDS) protein sample buffer containing 100 mM
Tris-HCl (pH 6.8), 200 mM DTT, 4 SDS, 0.4% bromophenol blue, and
20% glycerol. Protein was quantified using the Bradford Protein
assay kit II (Bio-Rad Laboratories, Inc.). Equal quantities of
protein (25 µg) were separated by 10% SDS-polyacrylamide gel
electrophoresis. The resolved proteins were then transferred to
polyvinyldifluoride membranes (Merck Millipore). The membranes were
blocked by incubation with 1% bovine serum albumin in TBS-Tween-20
(TBST; 10 mM Tris-HCl, 150 mM NaCl pH 7.5 containing 0.1% Tween-20)
at room temperature for 1 h and were then incubated with primary
antibodies against HIF1α (1:500), E-cadherin (1:200), vimentin
(1:500), Snail1 (1:200) and β-actin (1:1,000) for 1 h at room
temperature. The membranes were washed three times with TBST and
were then incubated for 30 min with goat anti-rabbit or goat
anti-mouse secondary antibody conjugated to alkaline phosphatase
(1:1,000; Santa Cruz Biotechnology, Inc.; cat. nos. sc-2007 and
sc-2008). The respective proteins were detected by colorimetric
reactions using 5-bromo-4-chloro-3′-indolyphosphate/nitro-blue
tetrazolium substrate.
Cell morphology and immunofluorescence
assay
Cells were collected at different time points (2–48
h) for analysis of cell morphological changes and at 48 h for
immunofluorescence and were fixed in 3.7% formaldehyde for 5 min.
Fixed cells were permeabilized in 0.1% Triton X-100 and were
incubated with antibodies specific to E-cadherin (1:200), vimentin
(1:100), and Snail1 (1:100) for 1 h at room temperature. The cells
were then washed and incubated with secondary antibodies for 30 min
at room temperature. Immunostaining was detected using the goat
anti-mouse and goat anti-rabbit secondary antibodies labeled with
Alexa Fluor 488 (1:500; Molecular Probes; Thermo Fisher Scientific,
Inc.; cat. nos. A11029 and A11034). Actin was stained using
rhodamine phalloidin (Thermo Fisher Scientific, Inc.). Cells were
examined using the Zeiss LSM 510 confocal imaging system (Carl
Zeiss, Oberkochen, Germany).
Statistical analysis
Data were analyzed using InStat statistics software
version 6.0 (GraphPad Software, Inc., La Jolla, CA, USA).
Statistical comparisons were performed using one-way analysis of
variance followed by Bonferroni post-hoc test for multiple
comparisons. P<0.05 was considered to indicate a statistically
significant difference.
Results
Dieckol attenuates
CoCl2-induced intracellular ROS levels in HT29
cells
Previous studies have suggested that hypoxia may
stimulate the generation of ROS in tumor cells, and increased ROS
promotes tumor progression; however, details of the underlying
molecular mechanism remain to be elucidated (10–13).
The investigation of ROS levels in tumor cells and antioxidants as
potential dietary supplements for the prevention of tumor
progression and potential cancer treatment has grown rapidly. The
present study used HT29 human colorectal cancer cells to assess the
association between hypoxia-induced intracellular ROS generation
and tumor progression. CoCl2 was used as a reagent to
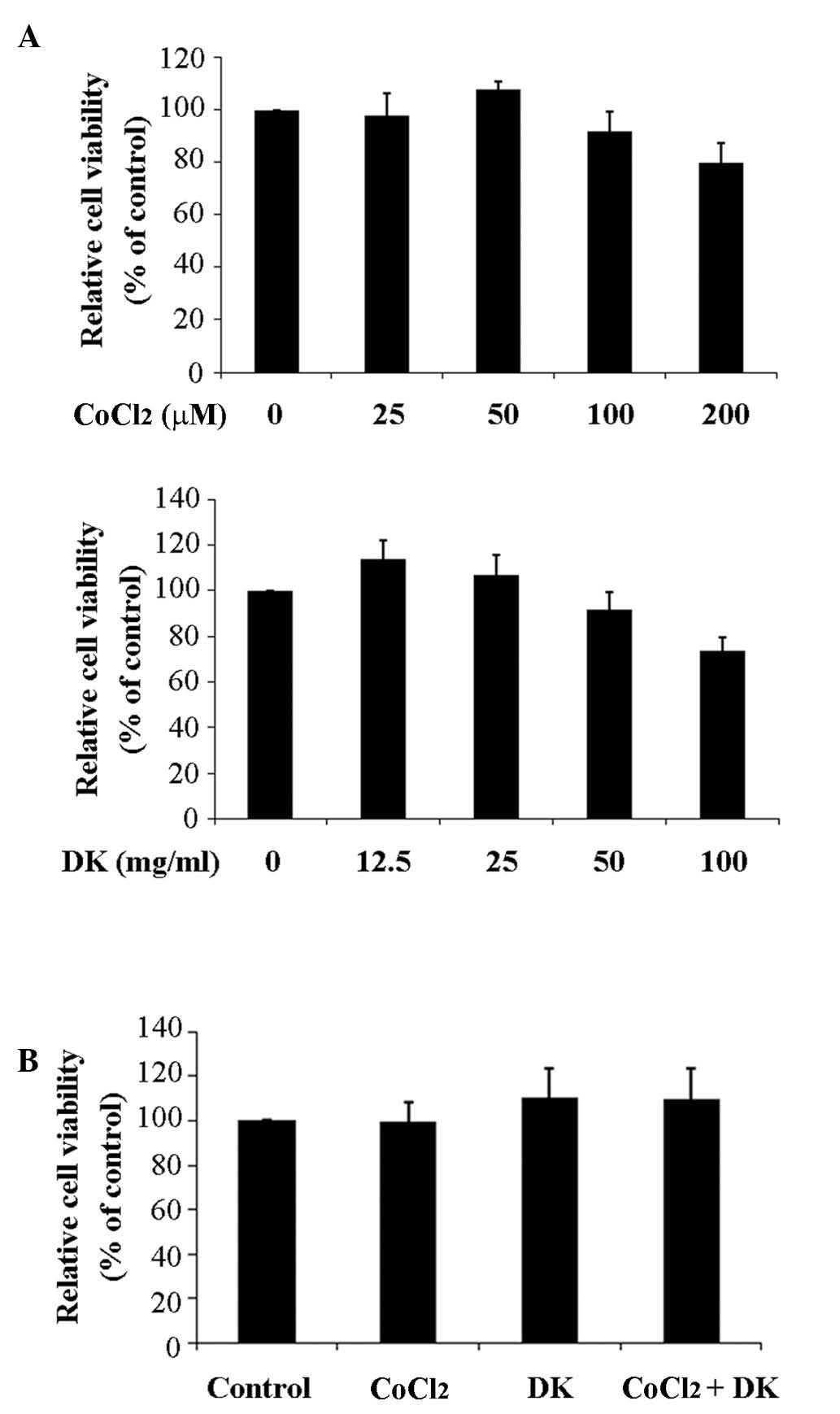
mimic the hypoxic microenvironment of tumor cells (8,15).
CoCl2 and dieckol did not affect the viability of HT29
cells at concentrations below 100 µM and 25 mg/ml (54 µM),
respectively (Fig. 1). As
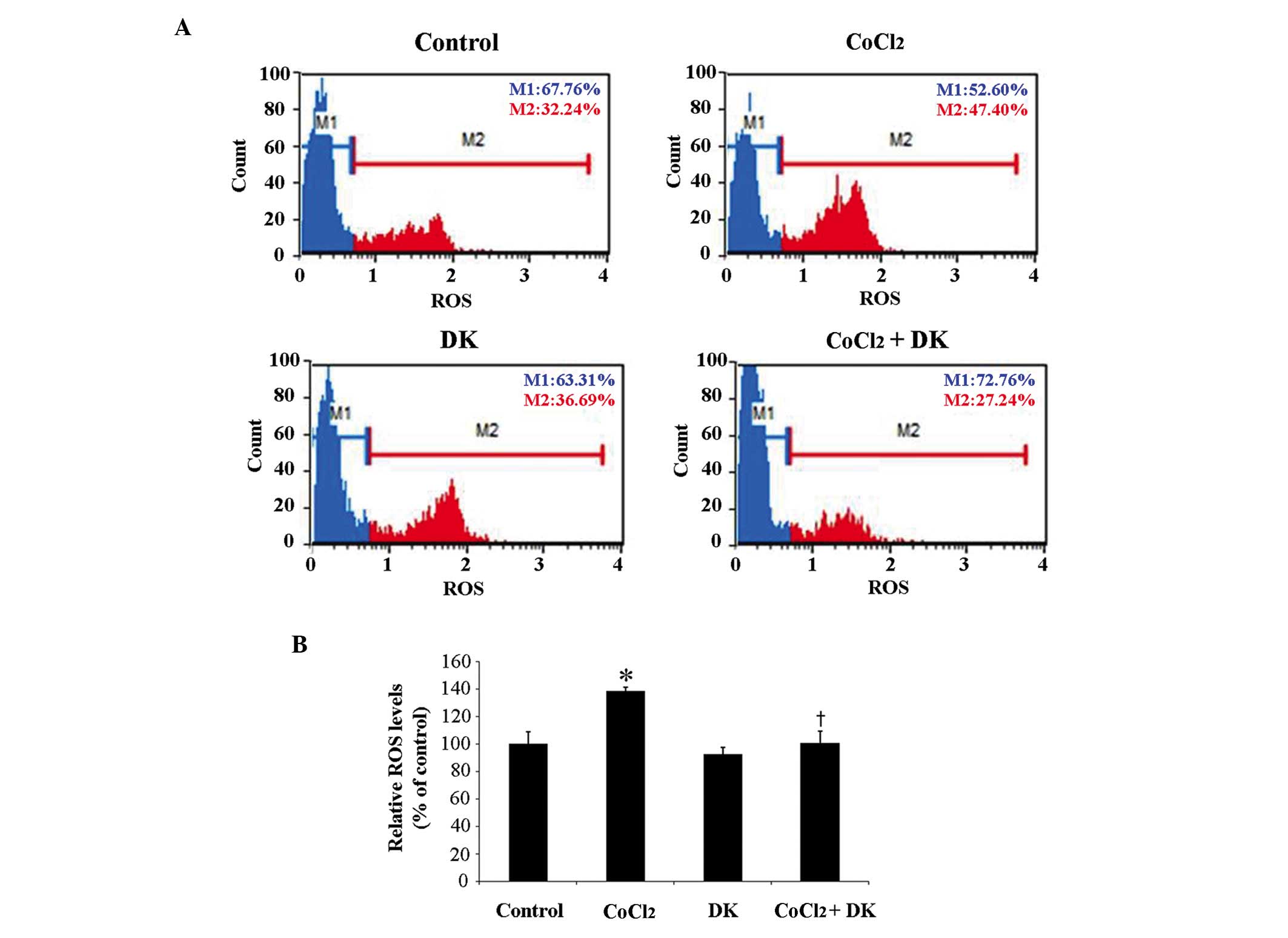
presented in Fig. 2, treatment of
HT29 cells with 50 µM CoCl2 resulted in significantly
increased intracellular ROS levels to ~140% compared with the
control (P<0.05; Fig. 2B),
suggesting that CoCl2 successfully mimicked the hypoxic
environment of tumor cells. In addition, in order to determine
whether intracellular ROS levels were associated with
hypoxia-induced tumor progression, HT29 cells were treated with 25
mg/ml dieckol in the absence and presence of 50 µM CoCl2
treatment. Dieckol treatment significantly attenuated the
CoCl2-induced increased cellular ROS levels (P<0.05;
Fig. 2B). These findings suggest
that HT29 cells may be responsive to CoCl2-induced
hypoxia and the antioxidant dieckol effectively reduced the
CoCl2-induced intracellular ROS levels.
Dieckol inhibits
CoCl2-induced EMT-associated phenotypic changes of HT29
cells
The effects of hypoxia-induced intracellular ROS on
the EMT of HT29 cells were also examined. EMT is characterized by
epithelial cells acquiring a mesenchymal appearance, such as the
loss of cell-cell contact, scattering from cell clusters,
acquisition of mesenchymal properties and the downregulation of
epithelium-specific proteins, such as E-cadherin, while
simultaneously expressing mesenchymal proteins such as vimentin and
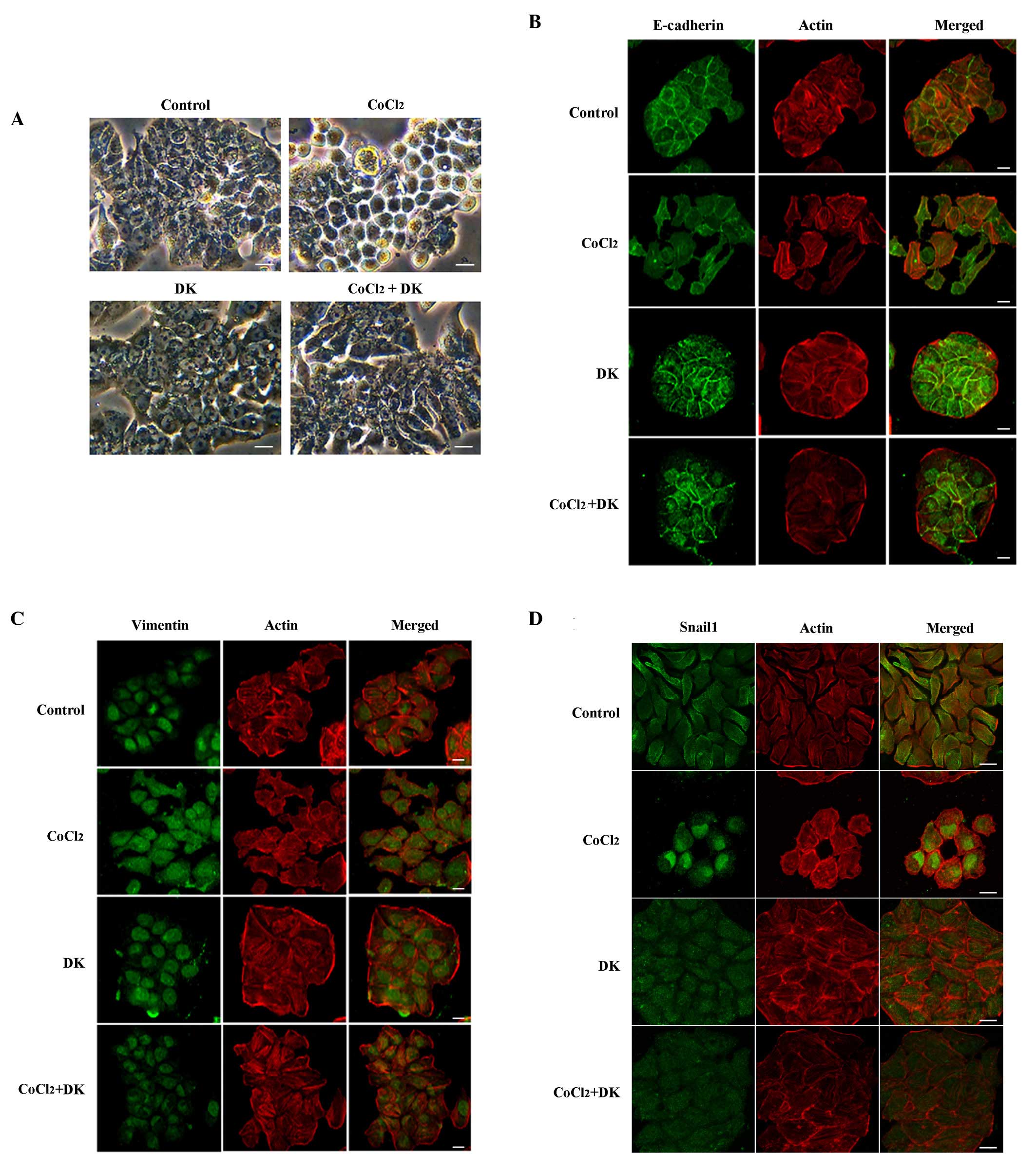
Snail1. As presented in Fig. 3A,
HT29 cells displayed typical epithelial characteristics with
well-developed cell-cell contact in the control group, without
CoCl2 treatment, whereas HT29 cells subjected to 50 µM
CoCl2 had reduced cell-cell contact and formed scattered
clusters. To examine whether the morphological changes in
CoCl2-treated HT29 cells were consistent with EMT,
immunofluorescence microscopy was performed, examining E-cadherin,
an epithelial cell marker, and vimentin and Snail1, mesenchymal
cell markers. As presented in Fig.
3B-D, E-cadherin signaling was primarily localized at the
cell-cell contact area in the control group (without
CoCl2 treatment). Following treatment with 50 µM
CoCl2 E-cadherin levels were decreased and diffused
throughout the cytoplasm, whereas nuclear vimentin signaling was
dispersed to the cytoplasm and membrane fraction. Nuclear
translocation of the transcription factor Snail1 was clearly
observed (Fig. 3D), suggesting
that the morphological changes observed in CoCl2-treated
HT29 cells may be identified as the onset of EMT. Cells were
treated with 50 µM CoCl2 in order to determine whether
CoCl2-induced intracellular ROS levels were associated
with EMT and subsequently incubated in the presence or absence of
25 mg/ml dieckol. Dieckol treatment inhibited the
CoCl2-induced mesenchymal morphologic changes and the
redistribution of E-cadherin, vimentin and Snail1, indicating that
the CoCl2-induced increase in ROS levels may be
associated with the occurrence of EMT in HT29 cells.
Dieckol decreases
CoCl2-induced migration and invasion of HT29 cells
EMT promotes the migration and invasion of cancer
cell (5–7). In order to investigate the effects of
CoCl2-induced hypoxia on migration and invasion of HT29
cells, the migration of HT29 cells was examined using Transwell
chambers. DMEM with 10% FBS was used as a chemoattractant in the
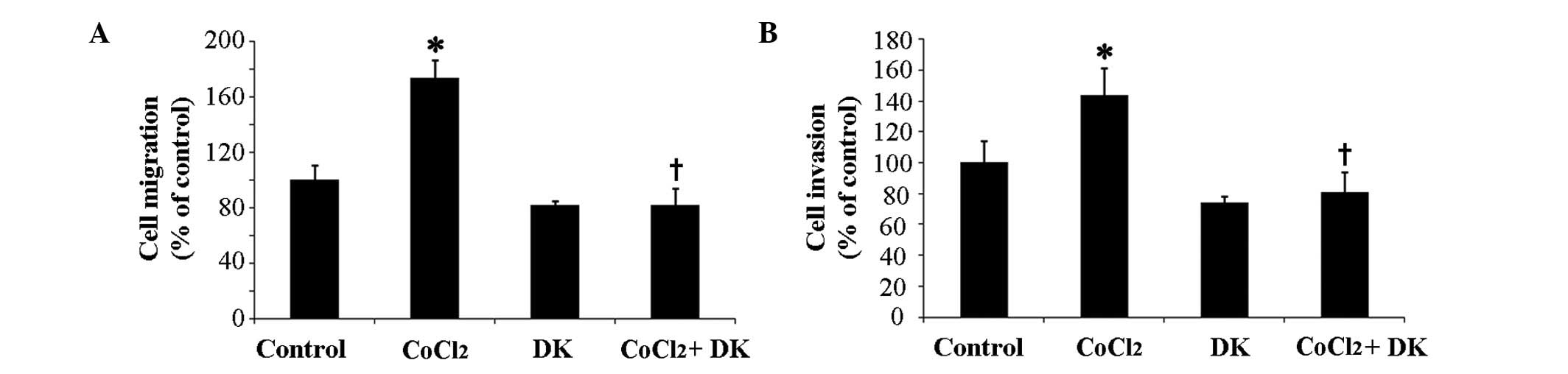
lower chamber. As presented in Fig.
4A, treatment with 50 µM CoCl2 significantly
increased migration of HT29 cells by ~1.7-fold compared with the
control (P<0.05), suggesting that CoCl2-induced
hypoxia may be associated with increased HT29 cell migration.
However, when HT29 cells were treated with 25 mg/ml dieckol
following incubation with CoCl2, the
CoCl2-induced increased migration of cells was
significantly inhibited (P<0.05; Fig. 4A). Invasion of HT29 cells on
reconstituted 3D Matrigel matrix significantly increased by
~1.4-fold in the presence of CoCl2 compared with the
control group (P<0.05; Fig. 4B)
and subsequent treatment with dieckol significantly inhibited the
CoCl2-induced invasive motility of cells (P<0.05;
Fig. 4B). These findings suggest
that the reduction of intracellular ROS levels following dieckol
treatment may decrease the hypoxia-induced migration and invasion
of HT29 cells.
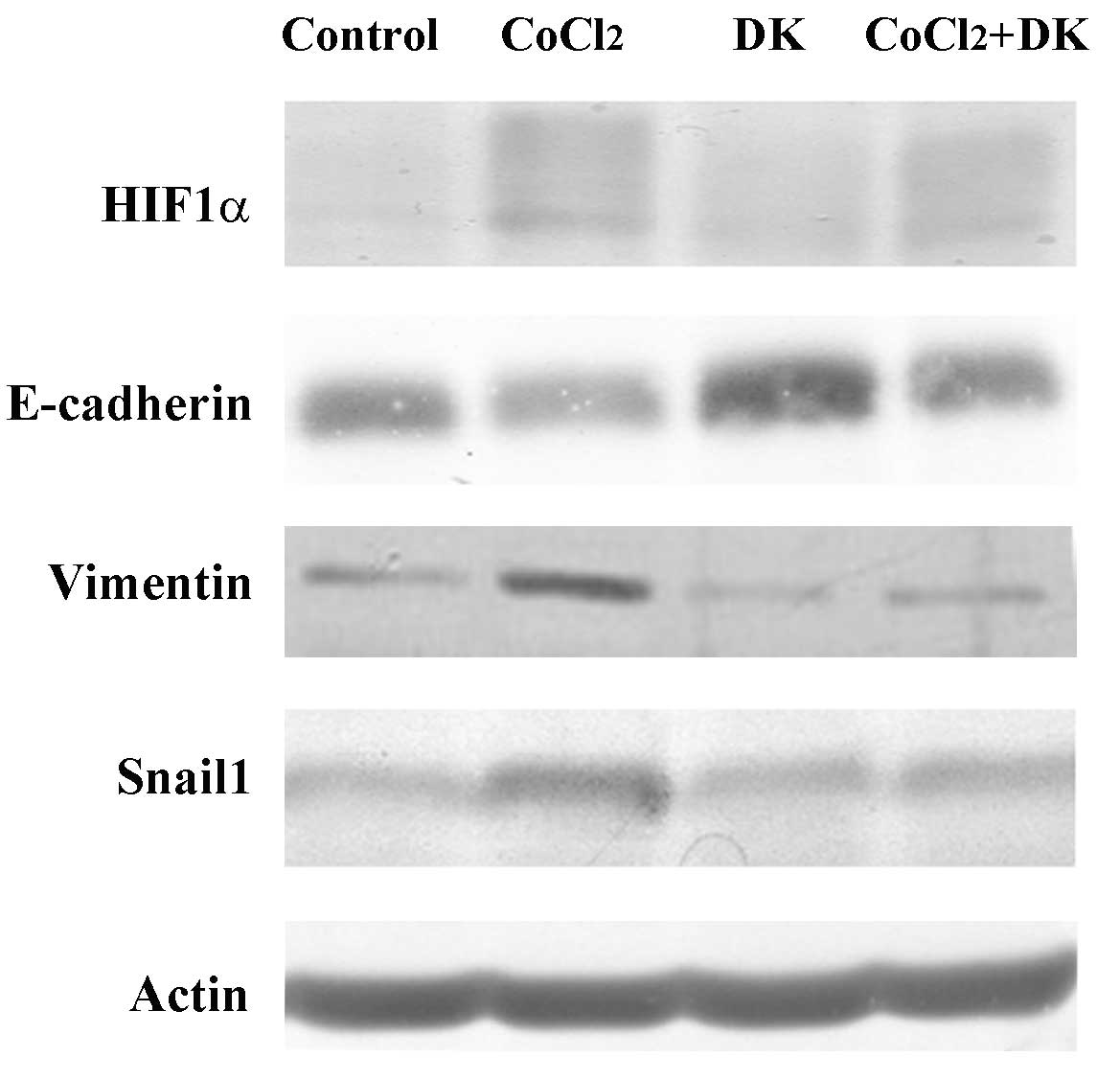
Dieckol suppresses the HIF1α signaling
pathway, required for CoCl2-induced EMT of HT29
cells
Subsequently, the CoCl2-induced
morphological changes and increased cell motility of HT29 cells
were verified by quantification of the changes in the intracellular
expression levels of EMT marker proteins. As aforementioned, EMT is
initially characterized by the downregulation of the epithelial
marker, E-cadherin, and by an upregulation of mesenchymal markers,
such as vimentin. In particular, under hypoxia-induced EMT,
selective transcription factors HIF1α and Snail1 are major factors
in regulating the expression of EMT marker proteins (8). Previous studies silenced HIF1α by RNA
interference (RNAi), which resulted in an increase in E-cadherin, a
decrease in vimentin and reduced cell invasion (16,17).
The HIF1α-dependent regulation of E-cadherin expression is induced
by upregulation of Snail1 (18,19).
Silencing of HIF1α reduced nuclear localization of Snail1, which
resulted in increased E-cadherin expression. In accordance with
these reports, when HT29 cells were cultured in 50 µM
CoCl2, HIF1α expression levels increased (Fig. 5). Expression levels of vimentin and
Snail1 were also increased, whereas the expression of E-cadherin
was decreased. Conversely, dieckol treatment reversed the
CoCl2-induced changes in expression levels of all EMT
marker proteins. These findings suggest that ROS levels and HIF1α
signaling are important for CoCl2-induced EMT in HT29
cells.
Discussion
Metastasis is required for tumor progression to
occur. Recent developments in early diagnosis and surgical
techniques have revealed that >90% of mortality in cancer
patients is due to the metastatic spread of primary tumors
(20). It is of note that in
patients with colorectal cancer, ~50% of patients with an early
diagnosis have already developed metastasis. Colorectal cancer may
metastasize to the liver, lung, peritoneum and various other
distant organs. The metastasis rates and mortality of colorectal
cancer have markedly increased, resulting in it becoming the third
most common type of cancer worldwide (21,22).
Conventional chemotherapy increases the survival rate of patients
with colorectal cancer; however, there is a wide range of acute and
long-term side effects that may substantially affect the quality of
life of patients. Non-toxic chemotherapeutic agents derived from
natural products may be used for future treatment of colorectal
cancer.
Dieckol is a natural and non-toxic polyphenol
compound and its antioxidant properties have previously been
examined (23,24). A previous study revealed the
anti-inflammatory and anticancer activities of dieckol are based on
the induction of apoptosis and inhibition of aberrant proliferation
by oxidative stress (14). The
present study demonstrated that dieckol inhibited the
hypoxia-induced metastatic properties of tumor cells by the
downregulation of intracellular ROS levels and reduction of HIF1α
signaling. CoCl2-induced hypoxia increased ROS
generation and promoted EMT in HT29 cells, subsequent treatment
with dieckol inhibited hypoxia-induced EMT.
Previous studies have reported that hypoxia-induced
EMT occurs downstream of HIF1α (25,26)
and hypoxia-induced intracellular ROS generation activates various
signaling components upstream of HIF1α such as phosphatidylinositol
3-kinase/AKT serine/threonine kinase 1 (27,28)
and mitogen-activated protein kinase (29–31).
Expression of the dominant negative HIF1α mutant, von Hippel-Lindau
tumor suppressor, or its silencing has been identified to increase
E-cadherin expression (16),
whereas E-cadherin RNAi increased the migration and invasion of
tumor cells in hypoxic conditions (18), indicating that a HIF1α-dependent
downregulation of E-cadherin may be a critical factor for the
occurrence of EMT during hypoxia. It has also been previously
reported that Snail1, another strong transcriptional repressor of
E-cadherin, may be regulated by interaction with HIF1α (18). Snail1 has been determined to be
overexpressed in various types of human cancer (31). The upregulation of Snail1 may
subsequently result in decreased E-cadherin levels and ultimately
EMT (18,32). HIF1α silencing has also been
identified to affect nuclear localization and activation of Snail1
and tumor cell invasion (19).
Therefore, it is possible that hypoxia-induced EMT-associated
events may be due to the activation of HIF1α and Snail in
conjunction with the reduction of E-cadherin. The findings of the
present study clearly supported this hypothesis as the association
between the expression of HIF1α and Snail1, E-cadherin and vimentin
in hypoxia-induced invasive tumor cell migration was evident.
In conclusion, the present study provided evidence
that hypoxia-induced EMT of HT29 human colorectal cancer cells may
be dependent on cellular ROS levels and the HIF1α signaling
pathway. In addition, the results suggested that the natural,
non-toxic antioxidant, dieckol, may be considered a novel
therapeutic agent for the treatment of metastatic colorectal
cancer.
Acknowledgements
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea,
funded by the Ministry of Education, Science and Technology (grant
nos. 2012R1A1A2041500 and 2015R1D1A3A01018506).
References
|
1
|
Krishnamachary B, Berg-Dixon S, Kelly B,
Agani F, Feldser D, Ferreira G, Iyer N, LaRusch J, Pak B, Taghavi P
and Semenza GL: Regulation of colon carcinoma cell invasion by
hypoxia-inducible factor 1. Cancer Res. 63:1138–1143.
2003.PubMed/NCBI
|
|
2
|
Koga F, Kihara K and Neckers L: Inhibition
of cancer invasion and metastasis by targeting the molecular
chaperone heat-shock protein 90. Anticancer Res. 29:797–807.
2009.PubMed/NCBI
|
|
3
|
Mendez O, Zavadil J, Esencay M, Lukyanov
Y, Santovasi D, Wang SC, Newcomb EW and Zagzag D: Knock down of
HIF-1alpha in glioma cells reduces migration in vitro and invasion
in vivo and impairs their ability to form tumor spheres. Mol
Cancer. 9:1332010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rankin EB and Giaccia AJ: The role of
hypoxia-inducible factors in tumorigenesis. Cell Death Differ.
15:678–685. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Giannoni E, Parri M and Chiarugi P: EMT
and oxidative stress: A bidirectional interplay affecting tumor
malignancy. Antioxid Redox Signal. 16:1248–1263. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tsai YP and Wu KJ: Hypoxia-regulated
target genes implicated in tumor metastasis. J Biomed Sci.
19:1022012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Luo Y, He DL, Ning L, Shen SL, Li L and Li
X: Hypoxia-inducible factor-1alpha induces the
epithelial-mesenchymal transition of human prostatecancer cells.
Chin Med J (Engl). 119:713–718. 2006.PubMed/NCBI
|
|
8
|
Zhang L, Huang G, Li X, Zhang Y, Jiang Y,
Shen J, Liu J, Wang Q, Zhu J, Feng X, et al: Hypoxia induces
epithelial-mesenchymal transition via activation of SNAI1 by
hypoxia-inducible factor −1α in hepatocellular carcinoma. BMC
Cancer. 13:1082013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang W, Wu Y, Yan Q, Ma F, Shi X, Zhao Y,
Peng Y, Wang J and Jiang B: Deferoxamine enhances cell migration
and invasion through promotion of HIF-1α expression and
epithelial-mesenchymal transition in colorectal cancer. Oncol Rep.
31:111–116. 2014.PubMed/NCBI
|
|
10
|
Liu LZ, Hu XW, Xia C, He J, Zhou Q, Shi X,
Fang J and Jiang BH: Reactive oxygen species regulate epidermal
growth factor-induced vascular endothelial growth factor and
hypoxia-inducible factor-1alpha expression through activation of
AKT and P70S6K1 in human ovarian cancer cells. Free Radic Biol Med.
41:1521–1533. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao JP, Zhou ZG, Hu HL, Guo Z, Wang T,
Zhen GH and Zhang ZX: The relationships among reactive oxygen
species, hypoxia-inducible factor 1alpha and cell proliferation in
rat pulmonary arterial smooth muscle cells under hypoxia. Sheng Li
Xue Bao. 59:319–324. 2007.PubMed/NCBI
|
|
12
|
Catalano V, Turdo A, Di Franco S, Dieli F,
Todaro M and Stassi G: Tumor and its microenvironment: A
synergistic interplay. Semin Cancer Biol. 23:522–532. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pettersen EO, Ebbesen P, Gieling RG,
Williams KJ, Dubois L, Lambin P, Ward C, Meehan J, Kunkler IH,
Langdon SP, et al: Targeting tumour hypoxia to prevent cancer
metastasis. from biology, biosensing and technology to drug
development: The METOXIA consortium. J Enzyme Inhib Med Chem.
30:689–721. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee SH, Park MH, Heo SJ, Kang SM, Ko SC,
Han JS and Jeon YJ: Dieckol isolated from Ecklonia cava inhibits
alpha-glucosidase and alpha-amylase in vitro and alleviates
postprandial hyperglycemia in streptozotocin-induced diabetic mice.
Food Chem Toxicol. 48:2633–2637. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Duan W, Chang Y, Li R, Xu Q, Lei J, Yin C,
Li T, Wu Y, Ma Q and Li X: Curcumin inhibits hypoxia inducible
factor-1α-induced epithelial-mesenchymal transition in HepG2
hepatocellular carcinoma cells. Mol Med Rep. 10:2505–2510.
2014.PubMed/NCBI
|
|
16
|
Krishnamachary B, Zagzag D, Nagasawa H,
Rainey K, Okuyama H, Baek JH and Semenza GL: Hypoxia-inducible
factor-1-dependent repression of E-cadherin in von hippel-lindau
tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A
and ZFHX1B. Cancer Res. 66:2725–2731. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chan DA and Giaccia AJ: Hypoxia, gene
expression and metastasis. Cancer Metastasis Rev. 26:333–339. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Evans AJ, Russell RC, Roche O, Burry TN,
Fish JE, Chow VW, Kim WY, Saravanan A, Maynard MA, Gervais ML, et
al: VHL promotes E2 box-dependent E-cadherin transcription by
HIF-mediated regulation of SIP1 and snail. Mol Cell Biol.
27:157–69. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cannito S, Novo E, Compagnone A, di Bonzo
L Valfre, Busletta C, Zamara E, Paternostro C, Povero D, Bandino A,
Bozzo F, et al: Redox mechanisms switch on hypoxia-dependent
epithelial-mesenchymal transition in cancer cells. Carcinogenesis.
29:2267–2278. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Christofori G: New signals from the
invasive front. Nature. 441:444–450. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Boyle P and Ferlay J: Mortality and
survival in breast and colorectal cancer. Nat Clin Pract Oncol.
2:424–425. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Naishadham D, Lansdorp-Vogelaar I, Siegel
R, Cokkinides V and Jemal A: State disparities in colorectal cancer
mortality patterns in the United States. Cancer Epidemiol
Biomarkers Prev. 20:1296–1302. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Park SJ and Jeon YJ: Dieckol from ecklonia
cava suppresses the migration and invasion of HT1080 cells by
inhibiting the focal adhesion kinase pathway downstream of Rac1-ROS
signaling. Mol Cells. 33:141–149. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ko SC, Lee M, Lee JH, Lee SH, Lim Y and
Jeon YJ: Dieckol, a phlorotannin isolated from a brown seaweed,
ecklonia cava, inhibits adipogenesis through AMP-activated protein
kinase (AMPK) activation in 3T3-L1 preadipocytes. Environ Toxicol
Pharmacol. 36:1253–1260. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Semenza GL: Hydroxylation of HIF-1: Oxygen
sensing at the molecular level. Physiology (Bethesda). 19:176–182.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dehne N, Fuhrmann D and Brüne B:
Hypoxia-inducible factor (HIF) in hormone signaling during health
and disease. Cardiovasc Hematol Agents Med Chem. 11:125–135. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Koshikawa N, Hayashi J, Nakagawara A and
Takenaga K: Reactive oxygen species-generating mitochondrial DNA
mutation up-regulates hypoxia-inducible factor-1alpha gene
transcription via phosphatidylinositol 3-kinase-Akt/protein kinase
C/histone deacetylase pathway. J Biol Chem. 284:33185–33194. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yan M, Rayoo M, Takano EA, KConFab
Investigators and Fox SB: BRCA1 tumours correlate with a HIF-1alpha
phenotype and have a poor prognosis through modulation of
hydroxylase enzyme profile expression. Br J Cancer. 101:1168–1174.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lan A, Liao X, Mo L, Yang C, Yang Z, Wang
X, Hu F, Chen P, Feng J, Zheng D and Xiao L: Hydrogen sulfide
protects against chemical hypoxia-induced injury by inhibiting
ROS-activated ERK1/2 and p38MAPK signaling pathways in PC12 cells.
PLoS One. 6:e259212011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yu D, Li M, Tian Y, Liu J and Shang J:
Luteolin inhibits ROS-activated MAPK pathway in myocardial
ischemia/reperfusion injury. Life Sci. 122:15–25. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang F, Liu S, Xi S, Yan L, Wang H, Song Y
and Sun G: Arsenic induces the expressions of angiogenesis-related
factors through PI3K and MAPK pathways in SV-HUC-1 human
uroepithelial cells. Toxicol Lett. 222:303–311. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Batlle E, Sancho E, Franci C, Dominguez D,
Monfar M, Baulida J and De Herreros A Garcia: The transcription
factor snail is a repressor of E-cadherin gene expression in
epithelial tumour cells. Nat Cell Biol. 2:84–89. 2000. View Article : Google Scholar : PubMed/NCBI
|