Introduction
Glioma is one of the common types of primary
intracranial tumor in humans (1).
Gliomas account for ~30% of all types of brain and central nervous
system tumor and 80% of all types of malignant brain tumor
(2). It often spreads via the
cerebrospinal fluid, metastasizes to the spinal cord and the normal
brain tissue, and forms a satellite tumor group around the primary
tumor due to its uncontrolled aggressive growth and invasion.
Despite developments in surgery, chemotherapy, radiotherapy and
combined treatment modalities, curing glioma completely remains a
challenge (3–5). The median survival rate is generally
<1 year due to the high rate of recurrence following surgery and
poor prognosis (3–6). Therefore, it is necessary to
investigate novel potential therapeutic agents and elucidate the
molecular mechanisms underlying their cytotoxic effect via clinical
investigations. Temozolomide (TMZ) is an imidazotetrazine
derivative of the alkylating agent. dacarbazine. TMZ exhibits
schedule-dependent antineoplastic activity by interfering with DNA
replication and demonstrates activity against recurrent glioma
(7,8). However, for reasons that remain to be
elucidated, clinical response is poor. Therefore, the exact effect
of TMZ on the malignancy of glioma cells and the underlying
mechanism require investigation.
The mitogen-activated protein kinase (MAPK)
signaling pathways are global regulators of cellular responses to
stress, which transduce signals through subsequent phosphorylation
events, culminating in the phosphorylation of the terminal MAPK and
altered cellular transcription profiles. There are three major MAPK
pathways, and the respective terminal MAPKs in these are p38, c-Jun
N-terminal kinase (JNK) and extracellular signal-regulated kinase
(ERK)1/2 or MAPK 42/44 (9). The
ERK pathway is activated by receptor tyrosine kinases through the
small G protein, Ras. The activation of Ras leads to the
phosphorylation of Raf, which in turn phosphorylates MAPK kinase
(MEK)1/2, a dedicated dual-specificity kinase that regulates the
phosphorylation of ERK1/2 by tyrosine and threonine residues
(10). Activated ERK1/2 can
translocate into the nucleus, where it stimulates numerous
transcription factors involved in cell survival, apoptosis,
differentiation and motility (11).
A number of molecules often exhibit abnormalities in
the pathogenesis of glioblastoma. For example, the upregulation
and/or constitutive activation of growth factors, including
epidermal growth factor (EGF), hepatocyte growth factor (HGF),
vascular endothelial growth factor (VEGF), and the growth factor
receptors, EGFR and HGFR, are often involved in the abnormal growth
and motility of glioma cells (12–16).
Certain signaling cascades, particularly MEK/ERK1/2 and
phosphoinositide 3-kinase/AKT are critical in the molecular
abnormalities. In addition, ERK1/2 exhibits constant constitutive
activation upon alterations of tyrosine kinase receptors in
glioblastoma (17–22). As anticancer drugs often affect
various signal transduction pathways, including those associated
with tumor growth, cell death and metastasis, targeting specific
signaling pathways is a strategy for the development of cancer
therapy (23–26). Thus, certain selective inhibitors
of pathways or molecules associated with the progression and
development of glioblastoma have been considered as molecular
targeting agents in cancer therapy. U0126, an ERK1/2-specific
inhibitor, can significantly inhibit the activation of ERK and
suppress ERK signaling (27–29).
Therefore, it may be necessary to use molecular inhibitors for
effective tumor treatment.
TMZ is a standard chemotherapeutic agent for the
treatment of glioblastoma multiforme, however, the exact effect of
TMZ on glioma remains to be fully elucidated In the present study,
the effects of TMZ on the growth and motility of glioma C6 cells
were investigated, and whether the ERK signaling pathway is
involved in its regulation was examined.
Materials and methods
Cell culture
The rat glioma C6 cbiell line was obtained from
American Type Culture Collection (Vanassas, MA, USA). The HEK 293T
cell line was stored in the Research Center for Vascular Biology
(Yangzhou University, Yangzhou, China), which was used to generate
adenoviral vectors. These cells were grown in Dulbecco's modified
Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 5% fetal bovine serum (FBS;
Lonza, Levallois-Perret, France) in a humidified atmosphere
containing 5% CO2 at 37°C.
Cell proliferation assay
Cell proliferation was assessed using an MTT assay.
The MTT assay was performed using a 96-well plate according to the
manufacturer's protocol. The cells were seeded at a density of
104 cells per well and were cultured at 37°C with 5% CO2
for 24 h, following which 250, 500 and 1,000 µM TMZ (Sigma-Aldrich;
Merck Millipore, Darmstadt, Germany) was added to the culture for
48 h or 500 µM TMZ for 24, 48 and 72 h. The cells were then
incubated with 20 µl of MTT (5 mg/ml in phosphate-buffered saline)
for 4 h at 37°C, following which the cells were lysed by the
addition of 200 µl dimethylsulfoxide. The absorbance was measured
at 570 nm using a Rainbow microplate reader (Tecan Austria GmbH,
Salzburg, Austria).
Migration and invasion assays
Cell migration was assessed using Transwell chambers
(6.5 mm; Corning Incorporated, Corning, NY, USA) with 8 µm pore
membranes. The lower chamber was filled with 600 µl DMEM medium
with or without TMZ. The cells (5×104) were suspended in
100 µl of DMEM medium with 1% FBS and plated into the upper
chamber, with or without 250, 500 and 1,000 µM TMZ for incubation
at 37°C. After 20 h, the number of cells visible on the
undersurface of the polycarbonate membranes following crystal
violet staining was scored in six randomly selected visual fields
(magnification, ×100) using a light microscope. For invasion
assays, the upper surface of the membrane was covered with 70 µl of
Matrigel (1 mg/ml; BD Biosciences, Franklin Lakes, NJ, USA). The
procedure of the invasion assay was the same as that for the
migration assay, with the exception that the incubation duration
was extended to 24 h.
Western blot analysis
The cells were lysed in cell lysis buffer for
western blot analysis and immunoprecipiation (cat. no. P0013;
Beyotime Institute of Biotechnology, Haimen, China) containing a
protease inhibitor cocktail (Roche Diagnostics, Branchburg, NJ,
USA). Cell lysate was centrifuged at 10,000 × g for 10 min and the
supernatant was collected. Protein samples were quantified using
BCA Protein Assay kit (Beyotime Institute of Biotechnology,
Jiangsu, China). The protein samples (50 µg) were separated by 12%
SDS-PAGE and transferred onto Immobilon-P membranes (EMD Millipore,
Billerica, MA, USA). The membranes were blocked with 5% BSA
(Sigma-Aldrich; Merck Millipore) in Tris-buffered saline Tween-20
(TBST) for 1 h, and incubated with specific primary antibodies
overnight at 4°C and then washed for 3 times with TBST, followed by
incubation with horseradish peroxidase (HRP)-conjugated secondary
antibodies for 2 h at room temperature. Anti-ERK rabbit polyclonal
antibody (1:1,000; cat. no. 9102), anti phosphorylated (p)-ERK pAb
(1:1,000; cat. no. 9101), anti-p-p38MAPK mouse monoclonal antibody
(1:1,000; cat. no. 5140), anti-p-JNK mouse (1:2,000; cat. no. 9255)
and anti-VEGF-C rabbit pAb (1:1,000; cat. no. 2445) were the
primary antibodies used for detection of target proteins, all
obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA).
HRP-conjugated goat anti-rabbit IgG secondary antibody (1:2,000,
cat. no. 7074), horse anti-mouse IgG (1:2,000, cat. no. 7076) were
used and were obtained from Cell Signaling Technology, Inc.
Enhanced chemiluminescence-detecting reagent (Amersham; GE
Healthcare Life Sciences, Chalfont, UK) was used for development.
GAPDH was probed using anti-GAPDH rabbit (1:1,000, cat. no. 2118,
Cell Signaling Technology, Inc.) as a loading control. The protein
blots were quantified by densitometry using QuantityOne software
version 4.5.0 (Bio-Rad Laboratories, Inc., Hercules, CA, USA), and
the quantity expressed was relative to the corresponding target
protein.
Expression of recombinant VEGF-C
The recombinant human VEGF-C was purchased from
R&D Systems, Inc. (Minneapolis, MN, USA). The VEGF C cDNA was
amplified by La Taq DNA polymerase (Takara Biotechnology Co., Ltd.,
Shanghai, China) in polymerase chain reaction (PCR) system with La
Taq buffer and 2.5 mM dNTP (Takara Biotechnology Co., Ltd.) using
the following primers: VEGF-C forward,
5′-AGTGTCAGGCAGCGAACAAGA-3′.and reverse,
5′-CTTCCTGAGCCAGGCATCTG-3′. PCR thermocycler 9700 (Applied
Biosystems) was used with the following regimen of thermal cycling:
stage 1, 1 cycle, 10 min at 95°C; stage 2, 40 cycles, 15 sec at
95°C, 1 min 60°C. The PCR products were cloned into a DL7001 Ad 5
adenoviral vector (Vector Core of Human Gene Therapy Institute,
University of Pennsylvania, Philadelphia, PA, USA). Answer: HEK
293T cells were transfected by adenoviral vector using
Lipofectamine 2000 (Invitrogen;Thermo Fisher Scientific, Inc.) in
cell culture dishes. The recombinant viruses were generated in HEK
293T cells and were used for infection of the glioma C6 cells at
37°C. After 24 h, the cells were used for the experiments. A
control cell line was constructed using the LacZ/Ad-5 control
vector (Vector Gene Technology Company Co., Ltd., Beijing,
China).
Statistic analysis
All the experiments were repeated at least three
times. Statistical significance was analyzed using the SPSS 11.0
software program (SPSS, Inc., Chicago, IL, USA). Data were analyzed
using Student's t-test. P<0.05 was considered to indicate a
statistically significant difference. Data are presented as the
mean ± standard error of the mean.
Results
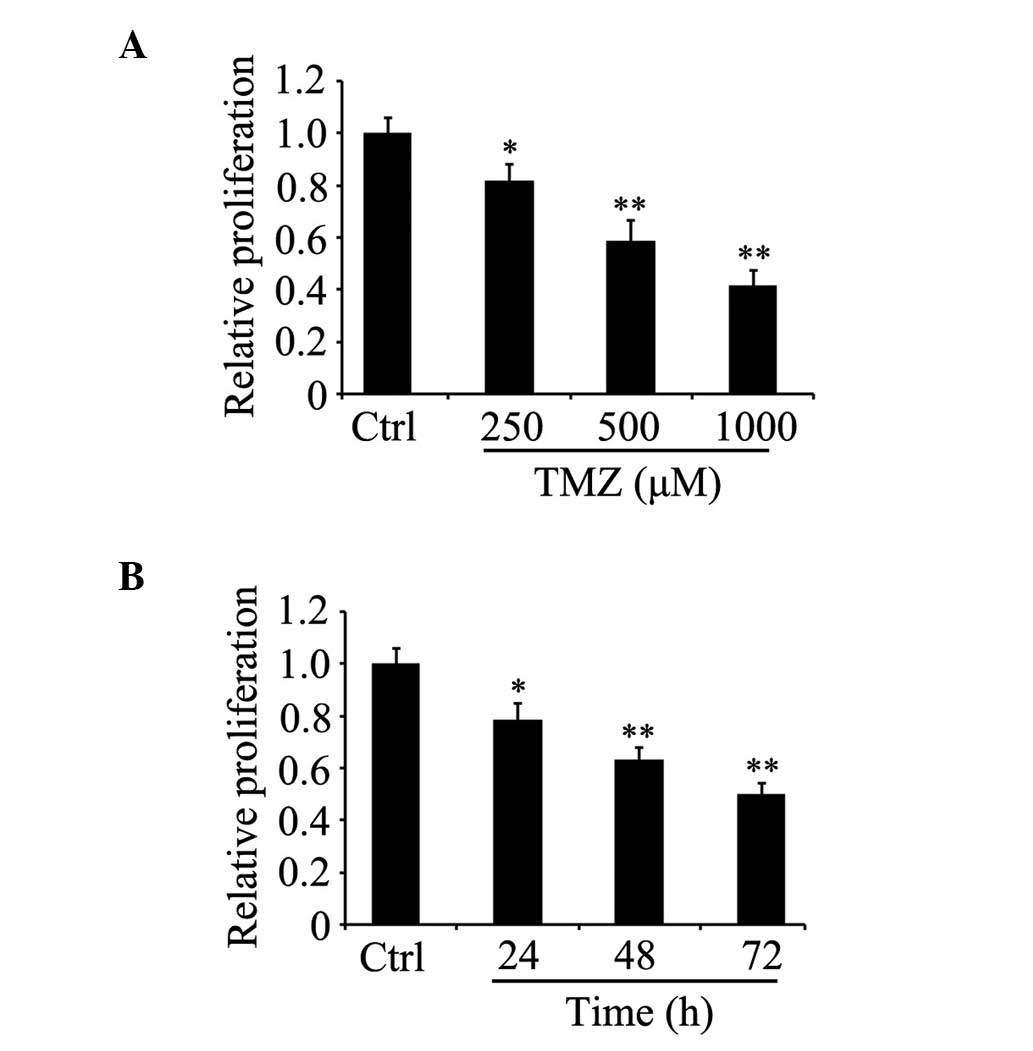
TMZ inhibits the proliferation of
glioma C6 cells
To determine whether TMZ affects cell viability, the
glioma C6 cells were grown in serum-containing medium for 24 h, and
were then either treated with TMZ at various concentrations for 48
h or were treated with 500 µM TMZ for different durations. An MTT
assay was then used for the analysis of cell proliferation. The
results indicated that the proliferation of the glioma C6 cells
treated with TMZ decreased in a concentration- and time-dependent
manner, compared with the untreated control cells (Fig. 1A and B).
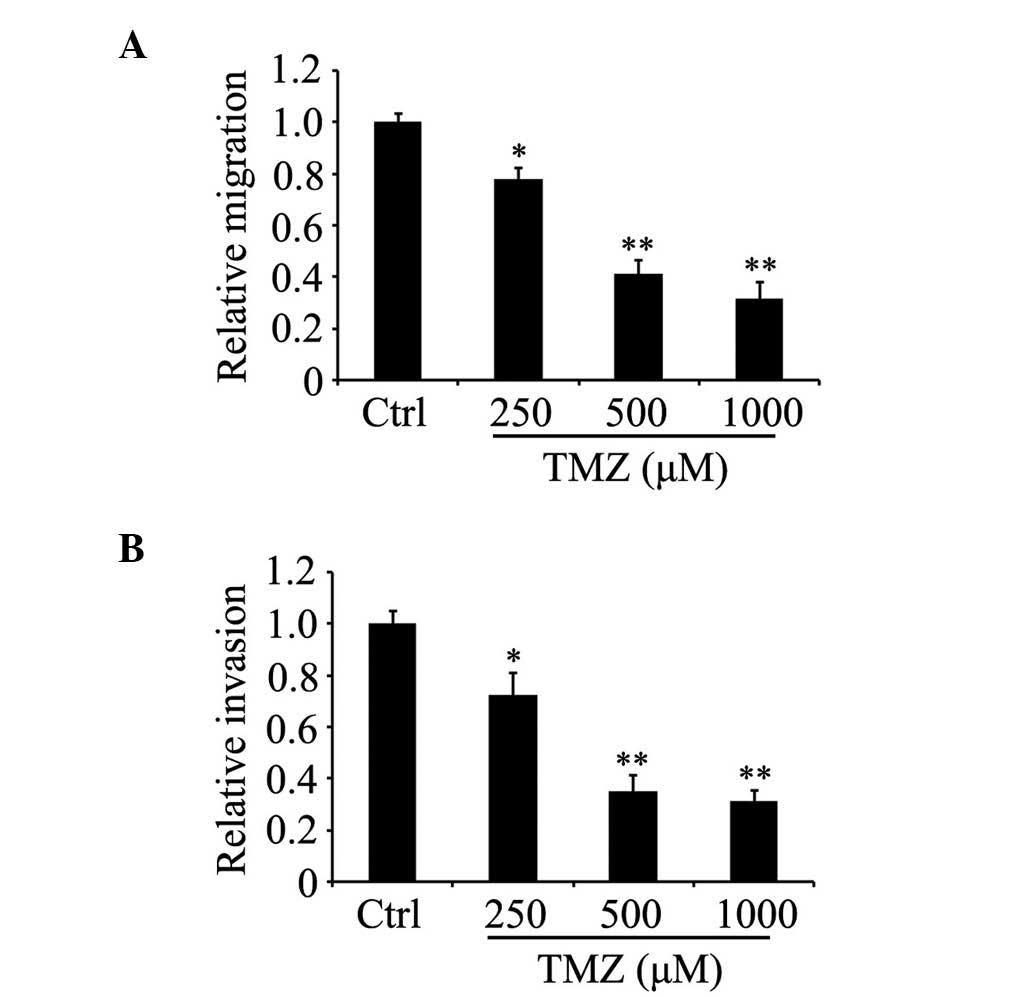
TMZ attenuates the migration and
invasion of glioma C6 cells
The effect of TMZ on glioma C6 cell motility was
determined by the detection of cell migration and invasion in
vitro. The Transwell assays indicated that the number of cells
able to migrate into the lower chambers decreased markedly in the
TMZ group as the concentration of TMZ gradually increased, compared
with the control group (Fig. 2A and
B). These data showed that TMZ significantly suppressed the
migration and invasion of glioma C6 cells in vitro in a
concentration-dependent manner.
TMZ decreases oncogenic phenotypes in
glioma C6 cells via MEK/ERK signaling
In order to clarify the molecular mechanism
underlying the effects of TMZ in regulating the growth and motility
of glioma C6 cells, the present study examined the activities of
members of the MAPK pathways, specifically the three terminal MAPK
molecules, p38, JNK and ERK, by detecting their phosphorylation.
The results showed that ERK1/2 phosphorylation was markedly
decreased in the TMZ-treated cells, compared with the untreated
control cells (Fig. 3A). However,
no significant differences were found between the TMZ-treated and
the untreated groups in the expression levels of p38 or JNK.
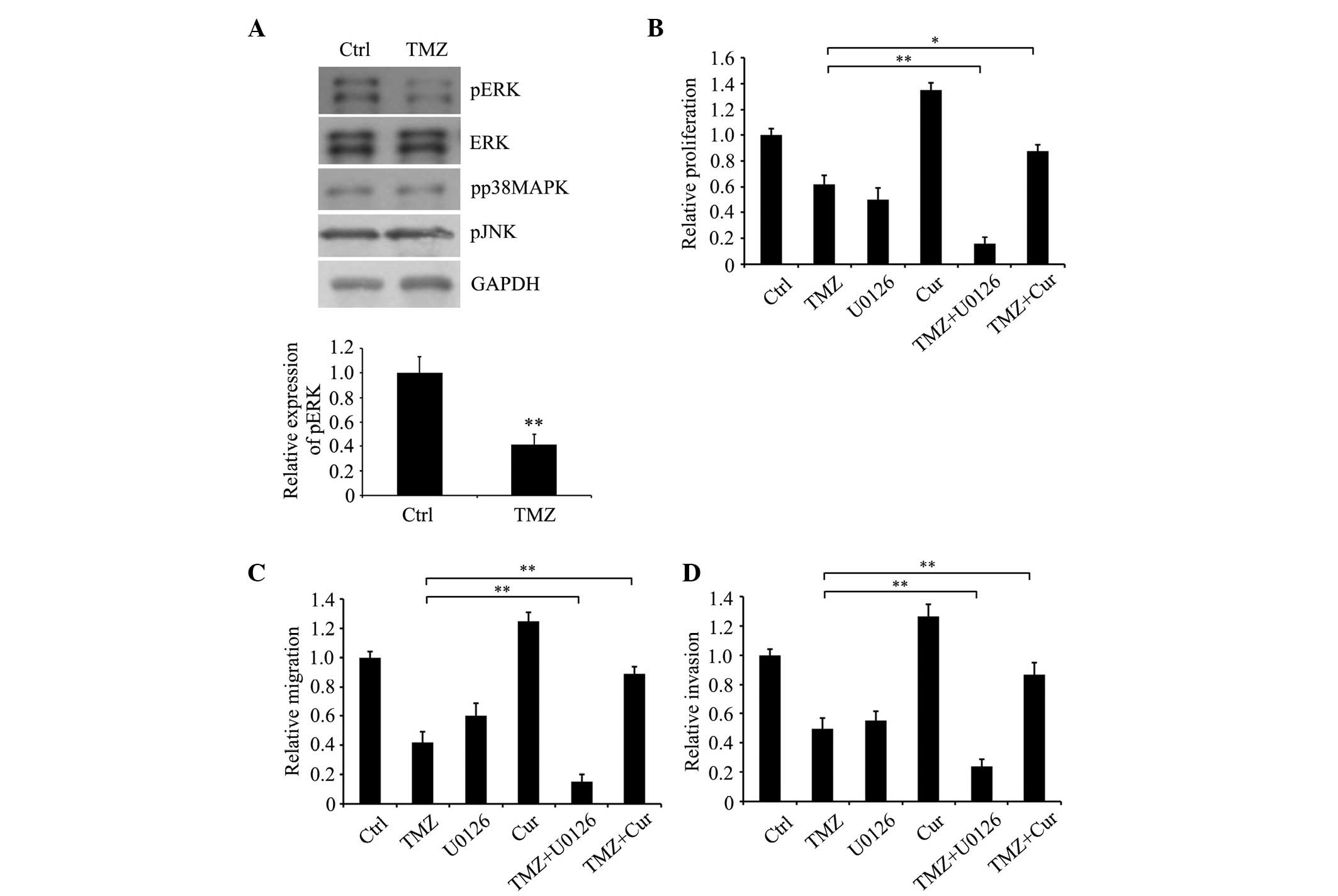 | Figure 3.TMZ attenuates the proliferation and
motility of glioma C6 cells via MAPK kinase/ERK signaling. (A)
Expression levels of pp38MAPK, pJNK, pERK, ERK and GAPDH were
detected in glioma C6 cells treated with or without 500 µM TMZ for
24 h using western blot analysis. (B) Proliferation, (C) migration
and (D) invasion of cells treated with TMZ following inhibition or
activation of ERK signaling by 30 nM U0126 and 1 µM Cur,
respectively. *P<0.05 and **P<0.01 (Student's t-test). Data
are presented as the mean ± standard error of the mean. TMZ,
temozolomide; Ctrl, untreated control cells; Cur, curcumin; ERK,
extracellular signal-regulated kinase; pERK, phosphorylated ERK;
pp38MAPK, phosphorylated p38 mitogen-activated protein kinase;
pJNK, phosphorylated c-Jun N-terminal kinase. |
To determine the effect of ERK on the function of
TMZ in inhibiting the malignant phenotype of glioma C6 cells, the
ERK1/2 specific inhibitor, U0126, and MEK activator, curcumin, were
used. The results indicated that U0126 significantly augmented the
inhibitory effect of TMZ on the proliferation (Fig. 3B), migration (Fig. 3C) and invasion (Fig. 3D) of the cells, whereas a low
concentration of curcumin (30)
attenuated the inhibitory effect of TMZ on the oncogenic phenotypes
of the glioma C6 cells. This suggested that MEK/ERK signaling was
important in the TMZ-induced inhibition of malignant
phenotypes.
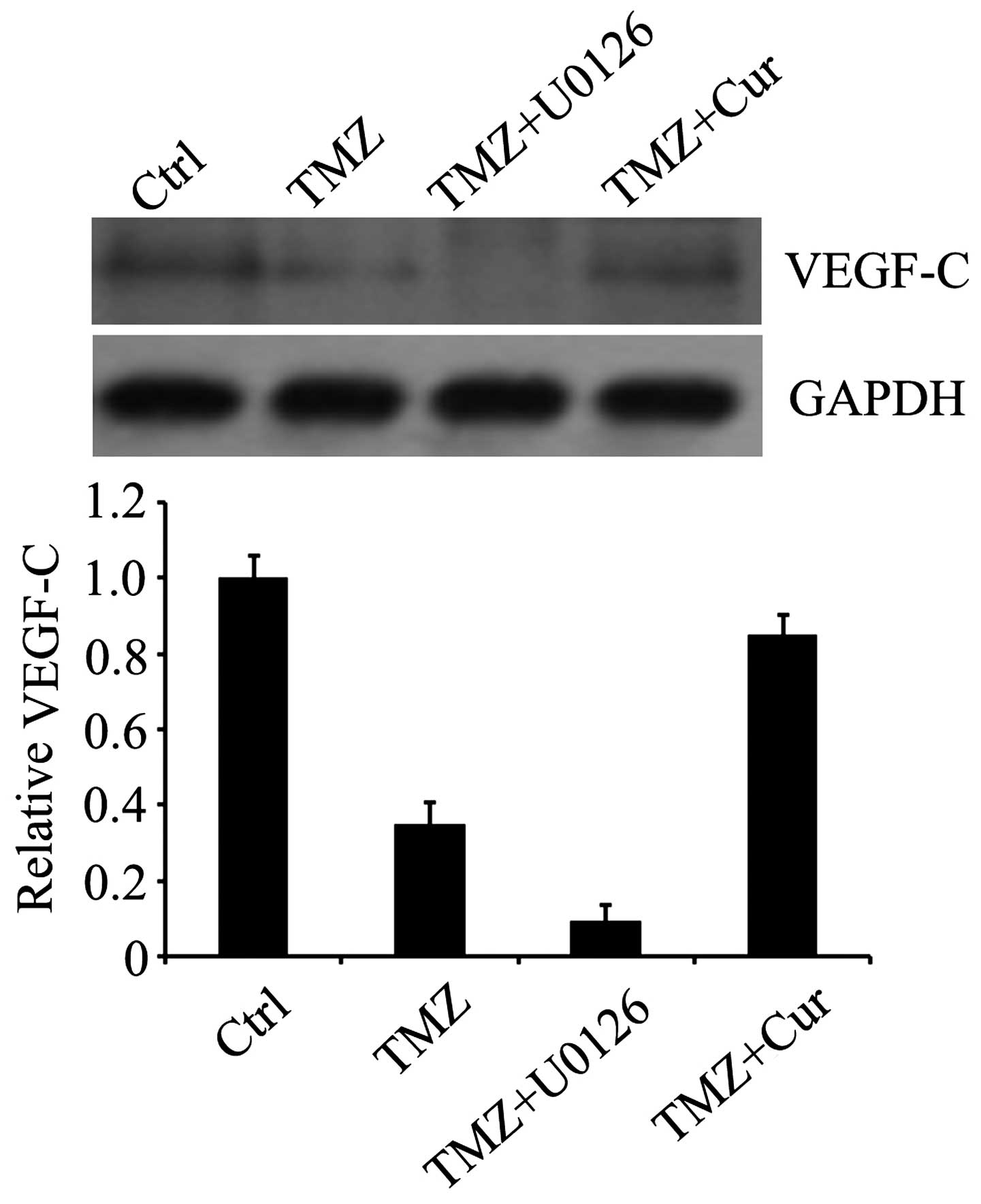
TMZ downregulates the expression of
VEGF-C via the ERK signaling pathway
As the upregulation of the growth factor, VEGF, is
often involved in the abnormal growth and metastasis of glioma
cells (13,15) and the expression of VEGF-C in
cancer cells can be affected by ERK signaling (31,32),
the present study investigated whether TMZ affected the expression
of VEGF-C via the ERK pathway. The results indicated that TMZ
decreased the expression of VEGF-C. Its expression was also reduced
by the ERK1/2 inhibitor, U0126, and was restored by curcumin
(Fig. 4), suggesting that VEGF-C
may be the downstream effector of ERK1/2.
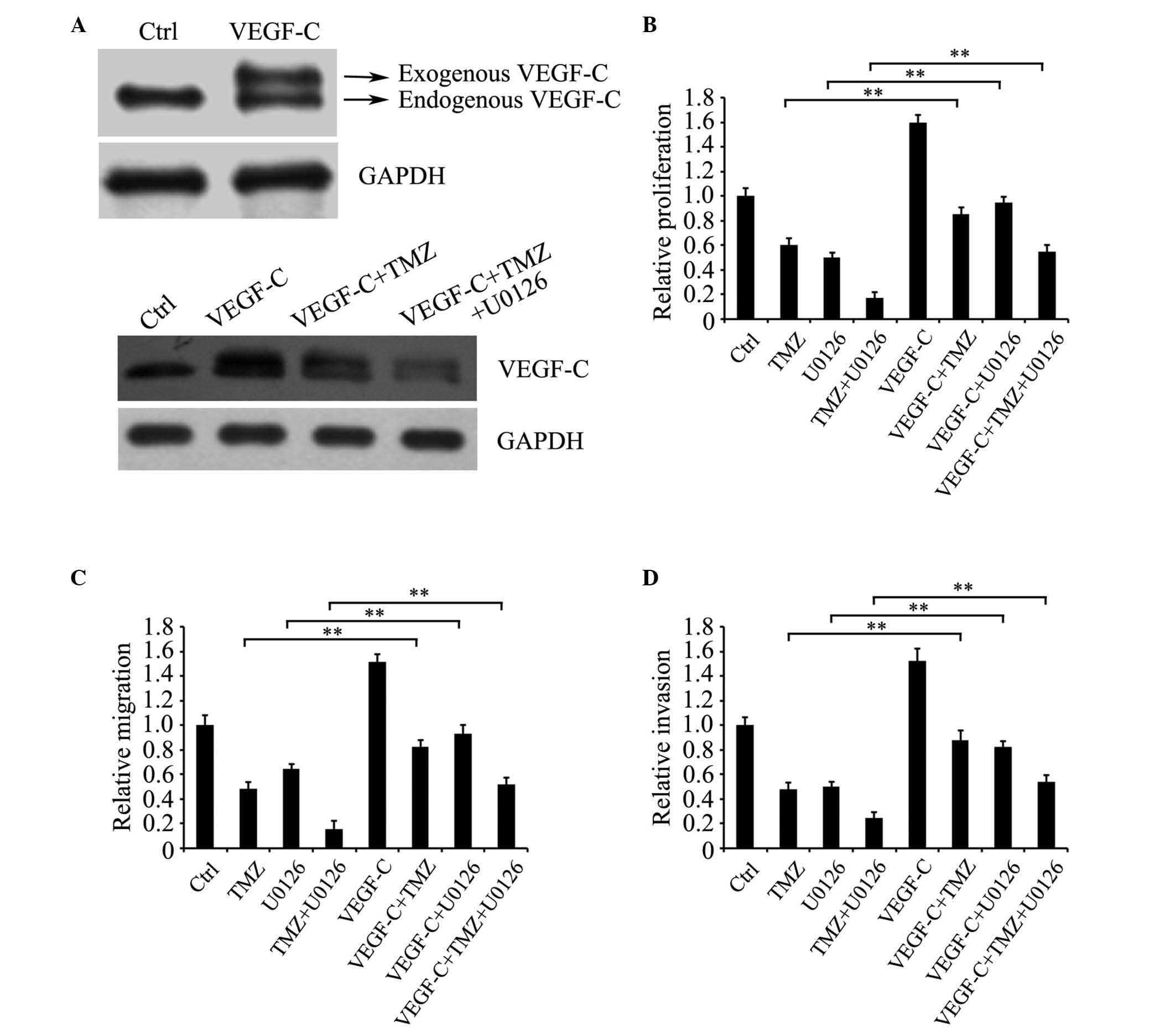
Overexpression of VEGF-C enhances the
growth and motility of TMZ-treated glioma C6 cells
To determine whether VEGF-C is involved in the
regulation of TMZ to affect glioma C6 cell growth and motility,
VEGF-C was overexpressed in glioma C6 cells and the cells were
treated with TMZ and/or U0126. Western blot analysis indicated that
the VEGF-C protein was markedly overexpressed in the glioma C6
cells, and treatment with TMZ and U0126 downregulated the protein
expression levels of exogenous and endogenous VEGF-C (Fig. 5A). The results of the MTT assay and
Transwell assay showed that the overexpression of VEGF-C
significantly reversed the growth, migration and invasion of the
TMZ- and/or U0126-treated cells, compared with the cells without
VEGF-C overexpression (Fig. 5B-D).
These data suggested that TMZ attenuated the malignancy of glioma
C6 cells, possibly through affecting the expression of VEGF-C via
ERK signaling.
Discussion
TMZ is a prodrug generated from the alkylating agent
dacarbazine by imidazotetrazine derivation. TMZ is often used as
standard therapy for glioma, based on its ability to methylate or
alkylate DNA at the O-6 or N-7 region of guanine residues leading
to the death of cancer cells (33,34).
However, the curative effect of TMZ is often compromised due to the
repair of DNA damage in tumor cells. Therefore, to use TMZ more
effectively, the mechanism underlying the inhibitory effects of TMZ
on tumor progression require further investigation. In the present
study, it was found that TMZ significantly inhibited the ERK
signaling pathway. The ERK pathway is a kinase cascade and is
critical in the biological functions of cancer cells (35–38).
In glioma cells, ERK signaling is closely associated with cell
death, senescence, proliferation, invasion and cellular
chemoresistance to TMZ (39–41).
The results of the present study indicated that the phosphorylation
of ERK1/2 was decreased in the TMZ-treated glioma C6 cells,
compared with the untreated control cells, and was accompanied by
decreased proliferation, migration and invasion. These decreases
were augmented by the ERK1/2 specific inhibitor, U0126, and were
attenuated by the MEK activator, curcumin, suggesting that TMZ
inhibited the oncogenic phenotypes of the glioma C6 cells, possibly
by inhibiting ERK signaling.
VEGF-C is a member of the VEGF family and is
involved in the malignancy of several types of tumor, including
those of colorectal, glioma, breast and prostate cancer (42–47).
It has been reported that VEGF-C can enhance cell growth, migration
and metastasis to promote tumor progression (48–52).
In the present study, the expression of VEGF-C was decreased in the
TMZ-treated glioma C6 cells, and overexpression of VEGF-C
attenuated the inhibitory effects of TMZ on cell proliferation,
migration and invasion. VEGF-C also enhanced cell growth and
motility, compared with the TMZ-treated cells without VEGF-C
overexpression, although they remained lower, compared with those
in the untreated control cells suggesting that there other
signaling pathways or proteins may be involved in the modulation.
The data obtained in the present study indicated that TMZ inhibited
tumor oncogenic phenotypes, including proliferation and motility,
at least in part, by downregulating the expression of VEGF-C in the
glioma C6 cells.
It has been reported that the expression of several
growth factors, including VEGF-C are regulated by ERK1/2 signaling
(31–53). In the present study, it was found
that TMZ downregulated ERK signaling and decreased the expression
of VEGF-C. It was hypothesized that TMZ decreases the expression of
VEGF-C through inhibiting the ERK signaling pathway in glioma C6
cells. As expected, the inhibition of ERK1/2 by its inhibitor,
U0126, decreased the expression of VEGF-C, whereas its expression
was upregulated by the MEK activator, curcumin, confirming the
hypothesis. However, the transcription factors involved in
modulating the expression of VEGF-C remain to be fully
elucidated.
In conclusion, the data obtained in the present
study indicated that ERK signaling was involved in the control of
TMZ on the oncogenic phenotypes of glioma C6 cells, and VEGF-C was
critical role in its modulation. In addition, the results suggests
that TMZ combined with ERK1/2 inhibitors may be a more effective
therapeutic approach in the treatment of glioma.
References
|
1
|
McDonald KL, O'Sullivan MG, Parkinson JF,
Shaw JM, Payne CA, Brewer JM, Young L, Reader DJ, Wheeler HT, Cook
RJ, et al: IQGAP1 and IGFBP2: Valuable biomarkers for determining
prognosis in glioma patients. J Neuropathol Exp Neurol. 66:405–417.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Goodenberger ML and Jenkins RB: Genetics
of adult glioma. Cancer Genet. 205:613–621. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang H, Ma L, Wang Q, Zheng X, Wu C and
Xu BN: Role of magnetic resonance spectroscopy for the
differentiation of recurrent glioma from radiation necrosis: A
systematic review and meta-analysis. Eur J Radiol. 83:2181–2189.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang X, Zhao HY, Zhang FC, Sun Y, Xiong ZY
and Jiang XB: Dendritic cell-based vaccine for the treatment of
malignant glioma: A systematic review. Cancer Invest. 32:451–457.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Balenci L, Clarke ID, Dirks PB, Assard N,
Ducray F, Jouvet A, Belin MF, Honnorat J and Baudier J: IQGAP1
protein specifies amplifying cancer cells in glioblastoma
multiforme. Cancer Res. 66:9074–9082. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Walbert T and Chasteen K: Palliative and
supportive care for glioma patients. Cancer Treat Res. 163:171–184.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Newlands ES, Stevens MF, Wedge SR,
Wheelhouse RT and Brock C: Temozolomide: A review of its discovery,
chemical properties, pre-clinical development and clinical trials.
Cancer Treat Rev. 23:35–61. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stevens MF, Hickman JA, Langdon SP, Chubb
D, Vickers L, Stone R, Baig G, Goddard C, Gibson NW, Slack JA, et
al: Antitumor activity and pharmacokinetics in mice of
8-carbamoyl-3-methyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one
(CCRG 81045; M & B 39831), a novel drug with potential as an
alternative to dacarbazine. Cancer Res. 47:5846–5852.
1987.PubMed/NCBI
|
|
9
|
Lewis TS, Shapiro PS and Ahn NG: Signal
transduction through MAP kinase cascades. Adv Cancer Res.
74:49–139. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rubinfeld H and Seger R: The ERK cascade:
A prototype of MAPK signaling. Mol Biotechnol. 31:151–174. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Luttrell LM: ‘Location, location,
location’: Activation and targeting of MAP kinases by G
protein-coupled receptors. J Mol Endocrinol. 30:117–126. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Auf G, Jabouille A, Delugin M, Guérit S,
Pineau R, North S, Platonova N, Maitre M, Favereaux A, Vajkoczy P,
et al: High epiregulin expression in human U87 glioma cells relies
on IRE1α and promotes autocrine growth through EGF receptor. BMC
Cancer. 13:5972013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Clara CA, Marie SK, de Almeida JR,
Wakamatsu A, Oba-Shinjo SM, Uno M, Neville M and Rosemberg S:
Angiogenesis and expression of PDGF-C, VEGF, CD105 and HIF-1α in
human glioblastoma. Neuropathology. 34:343–352. 2014.PubMed/NCBI
|
|
14
|
Li L, Puliyappadamba VT, Chakraborty S,
Rehman A, Vemireddy V, Saha D, Souza RF, Hatanpaa KJ, Koduru P,
Burma S, et al: EGFR wild type antagonizes EGFRvIII-mediated
activation of Met in glioblastoma. Oncogene. 34:129–134. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Burrell K, Singh S, Jalali S, Hill RP and
Zadeh G: VEGF regulates region-specific localization of
perivascular bone marrow-derived cells in Glioblastoma. Cancer Res.
74:3727–3739. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xie J, Ma YH, Wan M, Zhan RY and Zhou YQ:
Expression of dedifferentiation markers and multilineage markers in
U251 glioblastoma cells with silenced EGFR and FGFR genes. Oncol
Lett. 7:131–136. 2014.PubMed/NCBI
|
|
17
|
Zheng H, Ying H, Yan H, Kimmelman AC,
Hiller DJ, Chen AJ, Perry SR, Tonon G, Chu GC, Ding Z, et al: p53
and Pten control neural and glioma stem/progenitor cell renewal and
differentiation. Nature. 455:1129–1133. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Höland K, Boller D, Hagel C, Dolski S,
Treszl A, Pardo OE, Cwiek P, Salm F, Leni Z, Shepherd PR, et al:
Targeting class IA PI3K isoforms selectively impairs cell growth,
survival, and migration in glioblastoma. PLoS One. 9:e941322014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun Y, Zhang W, Chen D, Lv Y, Zheng J,
Lilljebjörn H, Ran L, Bao Z, Soneson C, Sjögren HO, et al: A glioma
classification scheme based on coexpression modules of EGFR and
PDGFRA. Proc Natl Acad Sci USA. 111:3538–3543. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee JS, Xiao J, Patel P, Schade J, Wang J,
Deneen B, Erdreich-Epstein A and Song HR: A novel tumor-promoting
role for nuclear factor IA in glioblastomas is mediated through
negative regulation of p53, p21, and PAI1. Neuro Oncol. 16:191–203.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Holland EC, Celestino J, Dai C, Schaefer
L, Sawaya RE and Fuller GN: Combined activation of Ras and Akt in
neural progenitors induces glioblastoma formation in mice. Nat
Genet. 25:55–57. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
McNamara MG, Sahebjam S and Mason WP:
Emerging biomarkers in glioblastoma. Cancers (Basel). 5:1103–1119.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rios A, Hsu SH, Blanco A, Buryanek J, Day
AL, McGuire MF and Brown RE: Durable response of glioblastoma to
adjuvant therapy consisting of temozolomide and a weekly dose of
AMD3100 (plerixafor), a CXCR4 inhibitor, together with lapatinib,
metformin and niacinamide. Oncoscience. 3:156–163. 2016.PubMed/NCBI
|
|
24
|
Berte N, Piee-Staffa A, Piecha N, Wang M,
Borgmann K, Kaina B and Nikolova T: Targeting homologous
recombination by pharmacological inhibitors enhances the killing
response of glioblastoma cells treated with alkylating drugs. Mol
Cancer Ther. 15:2665–2678. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shaaban S, Alsulami M, Arbab SA, Ara R,
Shankar A, Iskander A, Angara K, Jain M, Bagher-Ebadian H, Achyut
BR and Arbab AS: Targeting bone marrow to potentiate the anti-tumor
effect of tyrosine kinase inhibitor in preclinical rat model of
human glioblastoma. Int J Cancer Res. 12:69–81. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Qiu Z, Yue S, Chang J and Wang G:
Phosphatidylinositide 3-kinase inhibitor BKM120 suppresses
proliferation and promotes apoptosis of U251 glioblastoma cells. Xi
Bao Yu Fen Zi Mian Yi Xue Za Zhi. 32:936–939. 2016.PubMed/NCBI
|
|
27
|
Favata M, Horiuchi KY, Manos EJ, Daulerio
AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F,
et al: Identification of a novel inhibitor of mitogen-activated
protein kinase kinase. J Biol Chem. 273:18623–18632. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
DeSilva D, Jones EA, Favata MF, Jaffee BD,
Magolda RL, Trzaskos JM and Scherle PA: Inhibition of
mitogen-activated protein kinase kinase blocks T cell proliferation
but does not induce or prevent anergy. J Immunol. 160:4175–4181.
1998.PubMed/NCBI
|
|
29
|
Duncia JV, Santella JB, Higley CA, Pitts
WJ, Wityak J, Frietze WE, Rankin FW, Sun JH, Earl RA, Tabaka AC,
Teleha CA, et al: MEK inhibitors: The chemistry and biological
activity of U0126, its analogs, and cyclization products. Bioorg
Med Chem Lett. 8:2839–2844. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Son S, Kim KT, Cho DC, Kim HJ, Sung JK and
Bae JS: Curcumin stimulates proliferation of spinal cord neural
progenitor cells via a mitogen-activated protein kinase signaling
pathway. J Korean Neurosurg Soc. 56:1–4. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen X, Xie Q, Cheng X, Diao X, Cheng Y,
Liu J, Xie W, Chen Z and Zhu B: Role of interleukin-17 in
lymphangiogenesis in non-small-cell lung cancer: Enhanced
production of vascular endothelial growth factor C in
non-small-cell lung carcinoma cells. Cancer Sci. 101:2384–2390.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhu C, Qi X, Chen Y, Sun B, Dai Y and Gu
Y: PI3K/Akt and MAPK/ERK1/2 signaling pathways are involved in
IGF-1-induced VEGF-C upregulation in breast cancer. J Cancer Res
Clin Oncol. 137:1587–1594. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou X, Liao X, Zhang B, He H, Shui Y, Xu
W, Jiang C, Shen L and Wei Q: Recurrence patterns in patients with
high-grade glioma following temozolomide-based chemoradiotherapy.
Mol Clin Oncol. 5:289–294. 2016.PubMed/NCBI
|
|
34
|
Dong F: Metalloproteases involved in the
Temozolomide (TMZ) resistance of U87-MG glioma cells. Angenommen
vom Fachbereich Medizin der Philipps-Universität Marburg. April
3–2013.
|
|
35
|
Balmanno K and Cook SJ: Tumour cell
survival signalling by the ERK1/2 pathway. Cell Death Differ.
16:368–377. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M,
Tafuri A, et al: Roles of the Raf/MEK/ERK pathway in cell growth,
malignant transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Neuzillet C, Tijeras-Raballand A, de
Mestier L, Cros J, Faivre S and Raymond E: MEK in cancer and cancer
therapy. Pharmacol Ther. 141:160–171. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yoon S and Seger R: The extracellular
signal-regulated kinase: Multiple substrates regulate diverse
cellular functions. Growth Factors. 24:21–44. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gentile MT, Ciniglia C, Reccia MG,
Volpicelli F, Gatti M, Thellung S, Florio T, Melone MA and
Colucci-D'Amato L: Ruta graveolens L. induces death of glioblastoma
cells and neural progenitors, but not of neurons, via ERK 1/2 and
AKT activation. PLoS One. 10:e01188642015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu Q, Xu X, Zhao M, Wei Z, Li X, Zhang X,
Liu Z, Gong Y and Shao C: Berberine induces senescence of human
glioblastoma cells by downregulating the EGFR-MEK-ERK signaling
pathway. Mol Cancer Ther. 14:355–363. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Han S, Li Z, Master LM, Master ZW and Wu
A: Exogenous IGFBP-2 promotes proliferation, invasion, and
chemoresistance to temozolomidein glioma cells via the integrin
β1-ERK pathway. Br J Cancer. 111:1400–1409. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tacconi C, Correale C, Gandelli A,
Spinelli A, Dejana E, D'Alessio S and Danese S: Vascular
endothelial growth factor C disrupts the endothelial lymphatic
barrier to promote colorectal cancer invasion. Gastroenterology.
148:1438–1451.e8. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Carpenter RL, Paw I, Zhu H, Sirkisoon S,
Xing F, Watabe K, Debinski W and Lo HW: The gain-of-function GLI1
transcription factor TGLI1 enhances expression of VEGF-C and TEM7
to promote glioblastoma angiogenesis. Oncotarget. 6:22653–22665.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pang XH, Tian H, Liu ZY, Li SM, Liu ST and
Tian GP: Significance and expression of vascular endothelial growth
factor-C (VEGF-C) in esophageal squamous carcinoma and glioma. Ai
Zheng. 22:1166–1169. 2003.(In Chinese). PubMed/NCBI
|
|
45
|
Wang CA, Harrell JC, Iwanaga R, Jedlicka P
and Ford HL: Vascular endothelial growth factor C promotes breast
cancer progression via a novel antioxidant mechanism that involves
regulation of superoxide dismutase 3. Breast Cancer Res.
16:4622014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Varney ML and Singh RK: VEGF-C-VEGFR3/Flt4
axis regulates mammary tumor growth and metastasis in an autocrine
manner. Am J Cancer Res. 5:616–628. 2015.PubMed/NCBI
|
|
47
|
Sun GG, Wang YD, Cui DW, Cheng YJ and Hu
WN: EMP1 regulates caspase-9 and VEGFC expression and suppresses
prostate cancer cell proliferation and invasion. Tumour Biol.
35:3455–3462. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Khromova N, Kopnin P, Rybko V and Kopnin
BP: Downregulation of VEGF-C expression in lung and colon cancer
cells decelerates tumor growth and inhibits metastasis via multiple
mechanisms. Oncogene. 31:1389–1397. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chen Y, Jiang L, She F, Tang N, Wang X, Li
X, Han S and Zhu J: Vascular endothelial growth factor-C promotes
the growth and invasion of gallbladder cancer via an autocrine
mechanism. Mol Cell Biochem. 345:77–89. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Su JL, Chen PS, Chien MH, Chen PB, Chen
YH, Lai CC, Hung MC and Kuo ML: Further evidence for expression and
function of the VEGF-C/VEGFR-3 axis in cancer cells. Cancer Cell.
13:557–560. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Su JL, Yang PC, Shih JY, Yang CY, Wei LH,
Hsieh CY, Chou CH, Jeng YM, Wang MY, Chang KJ, et al: The
VEGF-C/Flt-4 axis promotes invasion and metastasis of cancer cells.
Cancer Cell. 9:209–223. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Timoshenko AV, Rastogi S and Lala PK:
Migration-promoting role of VEGF-C and VEGF-C binding receptors in
human breast cancer cells. British J Cancer. 97:1090–1098. 2007.
View Article : Google Scholar
|
|
53
|
Takahashi O, Komaki R, Smith PD,
Jürgensmeier JM, Ryan A, Bekele BN, Wistuba II, Jacoby JJ,
Korshunova MV, Biernacka A, et al: Combined MEK and VEGFR
inhibition in orthotopic human lung cancer models results in
enhanced inhibition of tumor angiogenesis, growth, and metastasis.
Clin Cancer Res. 18:1641–1654. 2012. View Article : Google Scholar : PubMed/NCBI
|