Introduction
Camurati-Engelmann disease (CED, OMIM: 131300), also
termed Engelmann disease or progressive diaphyseal dysplasia (PDD),
is a rare autosomal dominant bone disorder, with >300 reported
cases since the disease was first characterized in the 1920's
(1).
Patients with CED commonly present pain in
extremities, waddling gait, easy fatigability, muscle weakness
(2) and cortical thickening of the
diaphysis of the long bones. Sclerotic changes at the base of the
skull and pelvis are also frequently observed in the patients
(3,4). The genetic cause of CED has been
identified as a mutation in the transforming growth factor β1
(TGFβ1) gene (5,6). However, diagnosis of the disease is a
challenge for clinicians due to its extensive phenotypic variation
(7,8).
The present study aimed to identify a Chinese family
with rare bone abnormalities. The proband and relatives in the
family received comprehensive clinical evaluation and provided
venous blood samples for molecular genetic analysis. The clinical
and genetic data pointed to a positive diagnosis of CED for the
present family. The present study may provide references for
diagnosis of novel CED cases, as well as for prenatal
screening.
Materials and methods
Study subjects
The present study included a Chinese family that was
suspected of having CED (Fig. 1).
The proband was a visitor in the outpatient department at The Third
Xiangya Hospital Affiliated to Central South University (Changsha,
China). Written informed consent was received from all individuals
and the present study was approved by the Medical and Ethical
Committee of The Third Xiangya Hospital.
Clinic diagnosis
All individuals received a detailed clinical
evaluation at The Third Xiangya Hospital. The patients underwent
radiological examination, and serum biochemical tests to detect
thyroxine (T4), parathyroid hormone (PTH), thyroid stimulating
hormone (TSH) and procalcitonin (PCT) were performed in the
clinical laboratory of the hospital.
Molecular analysis
A total of 5 ml peripheral vein blood was drawn from
the proband and relatives. The genomic DNA was extracted using the
phenol-chloroform method, quantified using a UV spectrometer
(DU800; Beckman Coulter, Inc., Brea, CA, USA) and stored at −20°C
until use. Specific primers (Sunny Biotech Co., Ltd., Shanghai,
China) were designed using Primer3 software (version, 4.0.0;
http://primer3.ut.ee) for the amplification of
all exons and the exon/intron boundary sequences of TGFβ1
(Table I). PCR was run in a volume
of 20 µl containing the PrimeSTAR Max DNA polymerase and PCR buffer
(Takara Biotechnology Co., Ltd., Dalian, China) and specific
primers for 30 cycles at 95°C for 30 sec, 60°C for 30 sec and 72°C
for 30 sec on an ABI9700 thermal cycler (Applied Biosystems; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) using the genomic DNA as
a template. PCR products were purified by agarose electrophoresis,
followed by forward and backward sequencing. The sequences were
determined using the ABI 3730 DNA Analyzer (Applied Biosystems;
Thermo Fisher Scientific, Inc.) and blasted with reference
sequences from GenBank (National Center for Biotechnology
Information, National Institutes of Health, Bethesda, MD, USA).
 | Table I.Primer sequences for TGFβ1 gene exon
amplification. |
Table I.
Primer sequences for TGFβ1 gene exon
amplification.
| TGFβ1 Exon | Sequence (5′-3′) | Amplicon size
(bp) |
|---|
| Exon 1 |
| 427 |
|
Upstream |
CTCCTACCTTTTGCCGGGA |
|
|
Downstream |
CTCCTTGGCCTAGTAGTCGG |
|
| Exon 2 |
| 243 |
|
Upstream |
TCACCTCTGCCAAACCCCTG |
|
| Downstream |
ATGCCCTGACCTTCCTTCTG |
|
| Exon 3 |
| 383 |
|
Upstream |
TTTCTGGAAGCCTGTTTGGG |
|
|
Downstream |
AATCACTCAGGTTTCCATGCC |
|
| Exon 4 |
| 246 |
|
Upstream |
TGAGCTGCACTCTCAGAGTG |
|
|
Downstream |
AGTCAGGGGATAGGGGCAT |
|
| Exon 5 |
| 272 |
|
Upstream |
CCCTCTGAGAGTGTGTGTGT |
|
|
Downstream |
AGCACCTGGTCAGCAGATA |
|
| Exon 6 |
| 352 |
|
Upstream |
AGGGAGACCCAGATGGAGATAG |
|
|
Downstream |
CTTTCTCTCTCCTCTTCCTCCG |
|
| Exon 7 |
| 398 |
|
Upstream |
GGAGATGGGAAGAGGGGATG |
|
|
Downstream |
GGCACGGGTGTCCTTAAATAC |
|
Results
Pedigree analysis
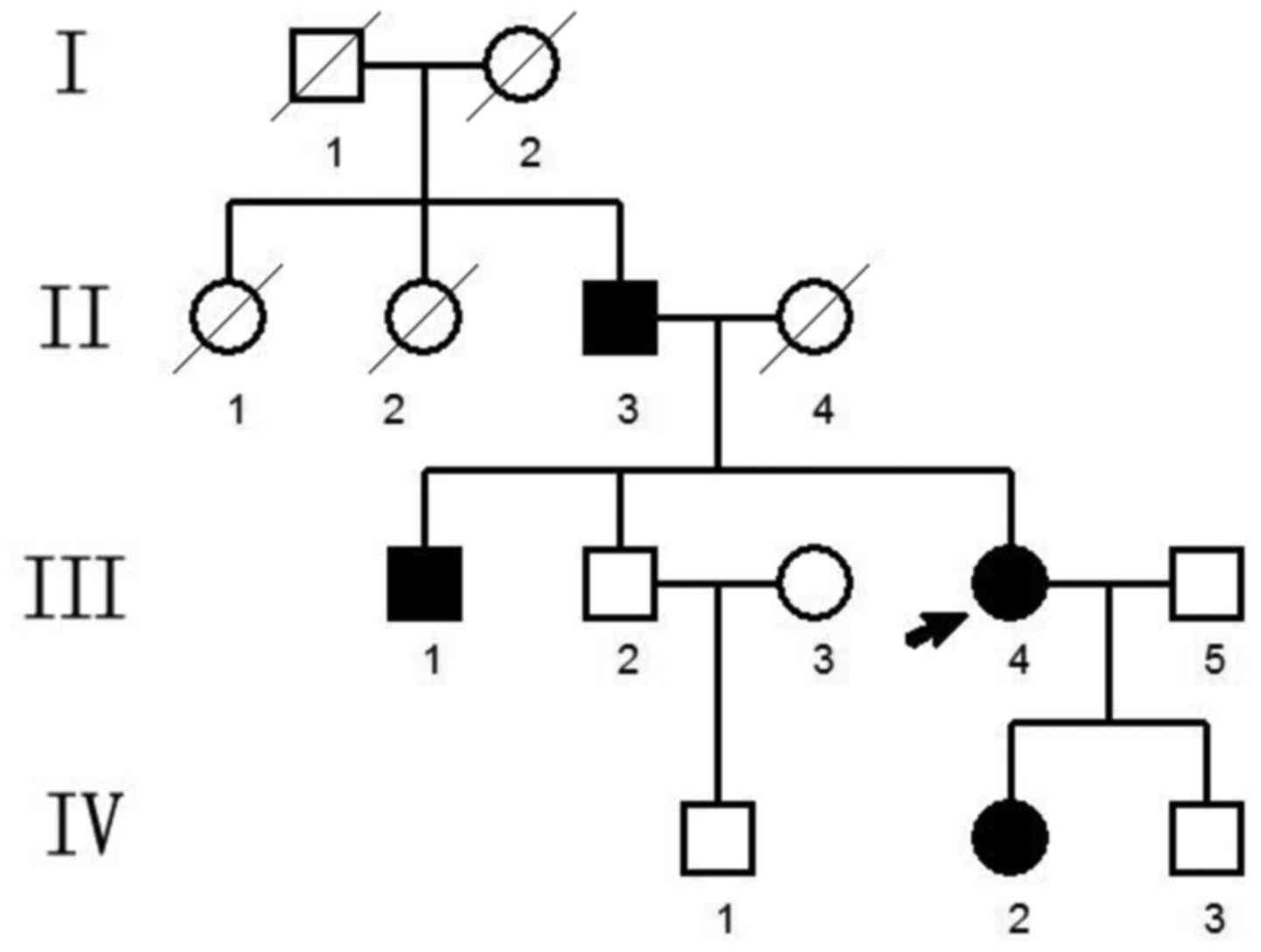
The pedigree covered four generations and 14
individuals in total (Fig. 1). A
total of four family members exhibited bone abnormalities, namely
the proband (III.4, female), and her father (II.3), brother (III.1)
and daughter (IV.2). This suggested a pattern of autosomal dominant
inheritance.
Clinic diagnosis
The clinical data from the patients are summarized
in Table II. All patients, with
the exception of IV.2, manifested pain in their extremities. All
patients in the family presented symptoms of waddling gait, muscle
weakness, skin tension over the affected bones and exophthalmos. No
patient exhibited acute pain syndrome, hepatosplenomegaly, cranial
nerve impairment, mental retardation, or a change to secondary
sexual characteristics. The proband, and patients II.3 and III.1
exhibited various symptoms, including dizziness, vision change and
hearing loss, which were not present in patient IV.2. The levels of
serum alkaline phosphatase (ALP), thyroid hormone and calcitonin
were in the normal range for all patients (ALP reference range for
males and females >15 years, 40–150 U/l; T4, 66–181 nmol/l; TSH,
0.27–4.2 µIU/ml; and PCT, <0.5 ng/ml). However, all patients
demonstrated a significant elevation in levels of serum PTH
(reference range, 15–65 pg/ml).
| Table II.Overview of patient clinical data. |
Table II.
Overview of patient clinical data.
|
| Patient |
|---|
|
|
|
|---|
| Characteristic | II3 | III1 | III4 | IV2 |
|---|
| Gender/Age
(years) | M/78 | M/47 | F/42 | F/17 |
| Pain in
extremities | + | + | + | − |
| Acute pain
syndrome | − | − | − | − |
| Waddling gait | + | + | + | + |
| Muscle weakness | + | + | + | + |
| Skin tension over
affected bones | + | + | + | + |
|
Hepatosplenomegaly | − | − | − | − |
| Cranial nerve
impairment | − | − | − | − |
| Metal
retardation | − | − | − | − |
| Dizziness | + | + | + | − |
| Vision change | + | + | + | − |
| Hearing loss | + | + | + | − |
| Exophthalmos | + | + | + | + |
| Secondary sexual
characteristics change | − | − | − | − |
| Serum alkaline
phosphatase elevation | − | − | − | − |
| Parathyroid hormone
elevation | + | + | + | + |
| Thyroid hormone
elevation | − | − | − | − |
| Calcitonin
elevation | − | − | − | − |
Radiological findings
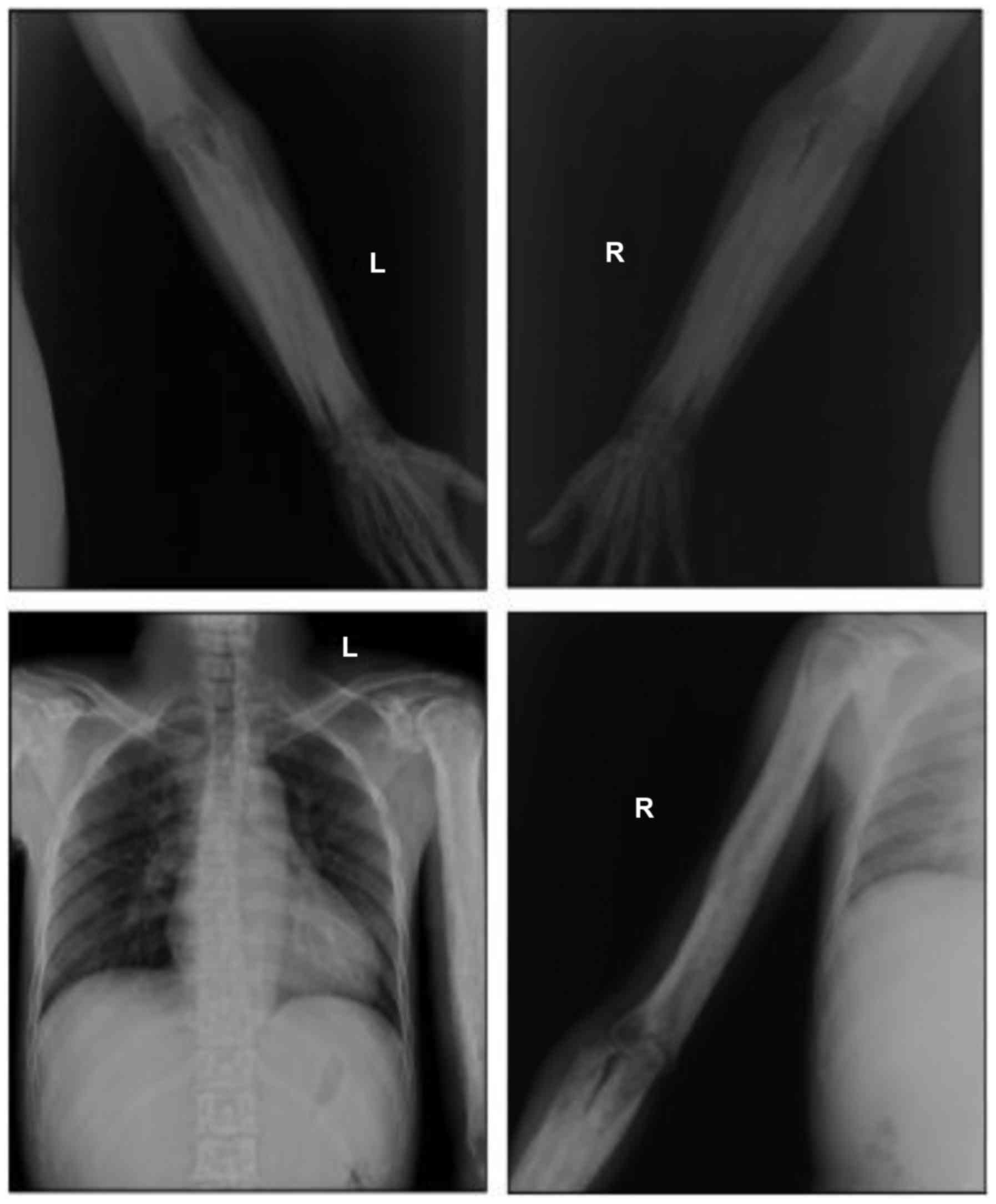
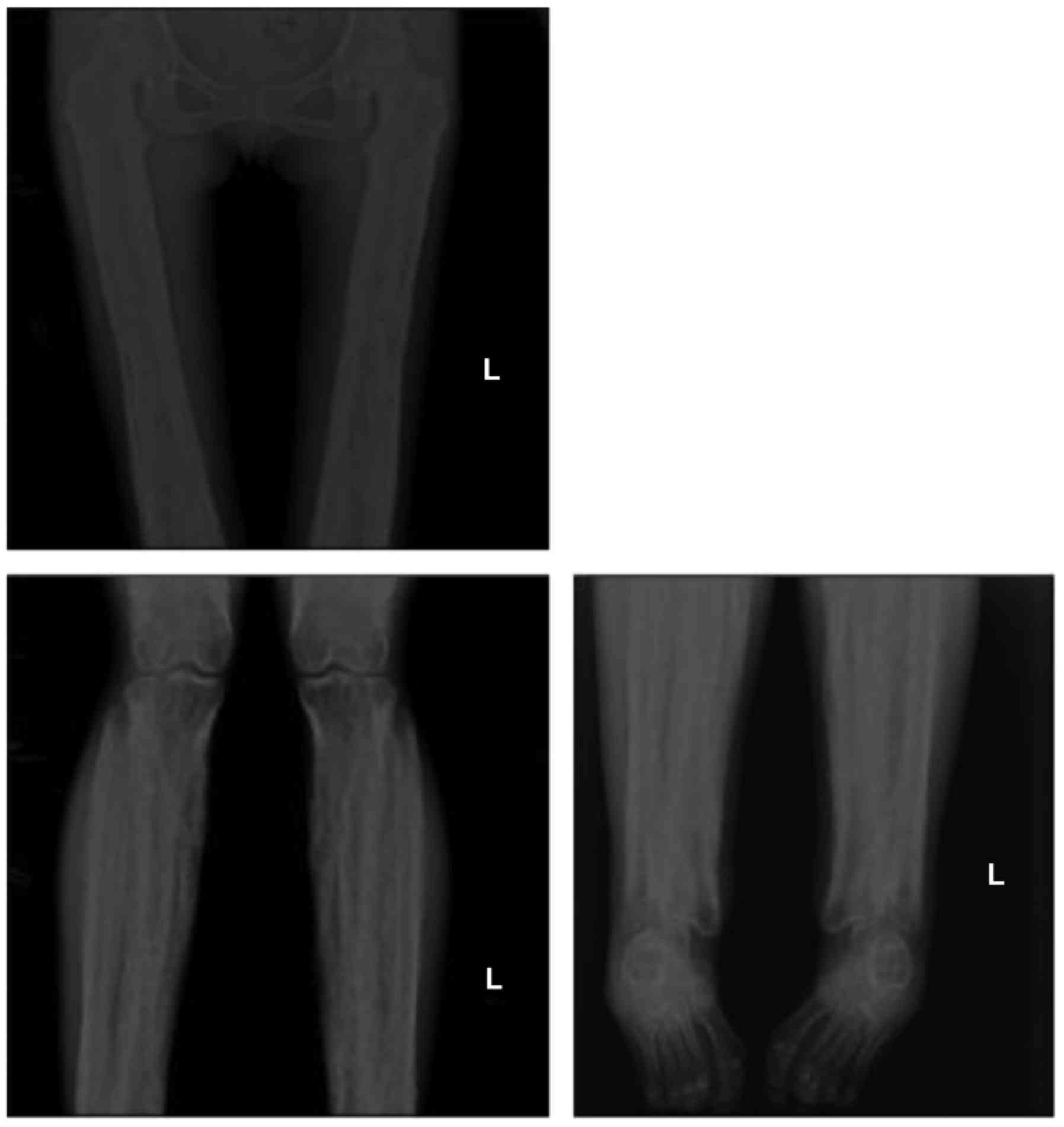
Radiographic images demonstrated cortical thickening
of the diaphysis of long bones with irregular endosteal sclerosis.
The bones involved included the humerus, radius, ulna, femur, tibia
and fibula. The joint space between radius and ulna was narrowed in
the patients, as well as the joint space of the tibia and fibula.
The metaphysis and epiphysis of the long bones were unaffected
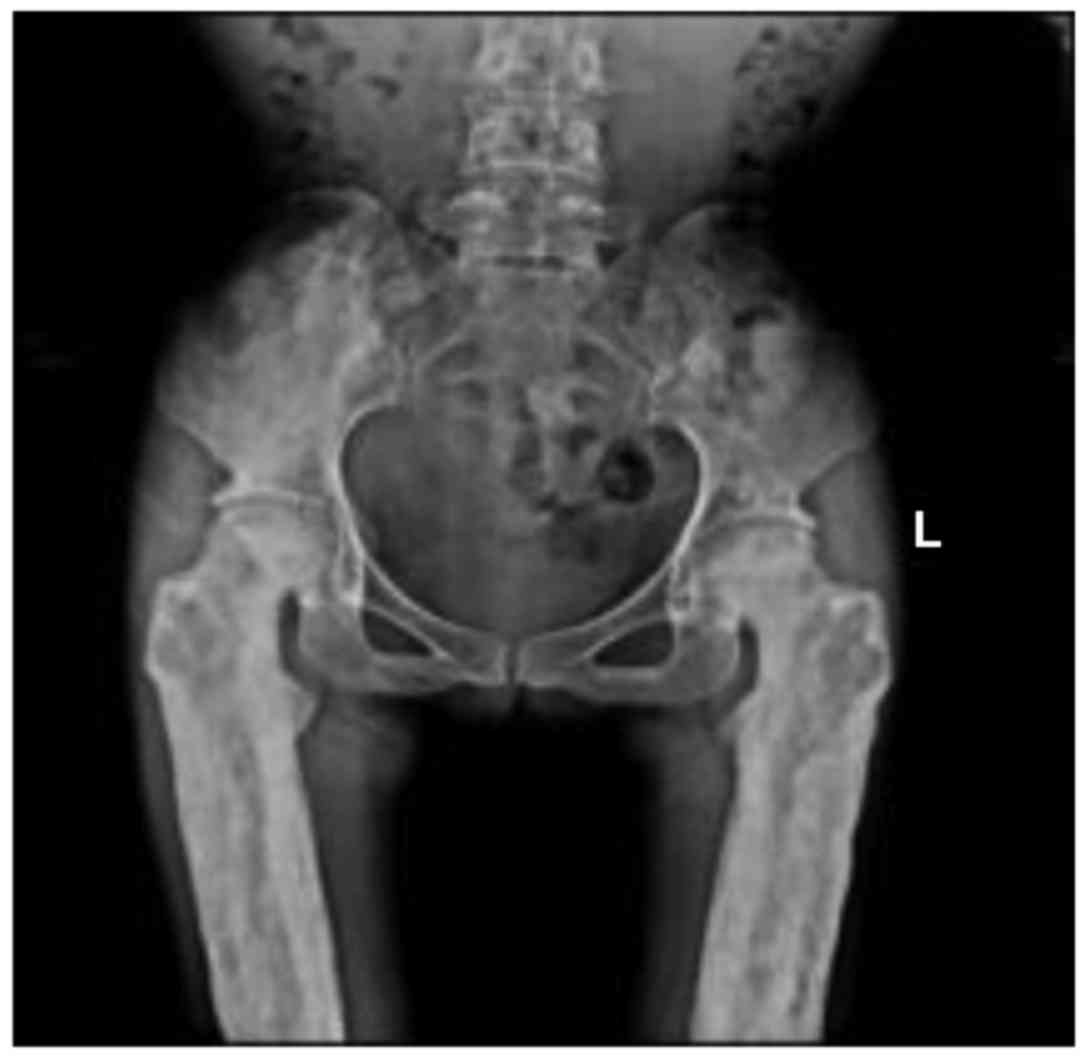
(Figs. 2 and 3). Osteoproliferation and osteosclerosis
of the ilium, acetabulum and ramus ossis ischii was observed
(Fig. 4). The images also revealed
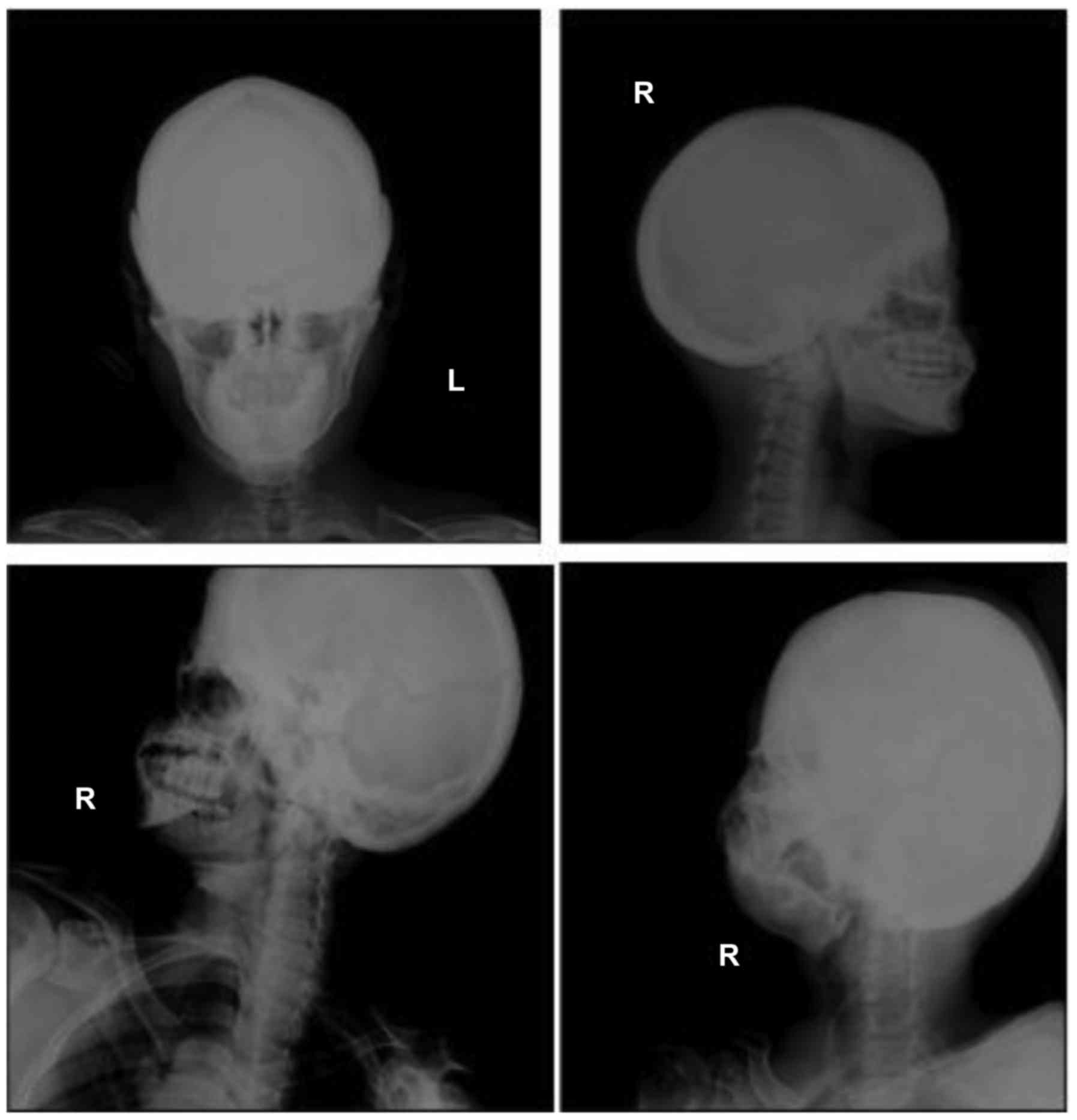
periosteal thickening and sclerosis of the skull and facial bone.
The diploe of skull disappeared, and the mastoid and sella turcica
presented cortical sclerosis (Fig.
5).
Identification of TGFβ1 mutations
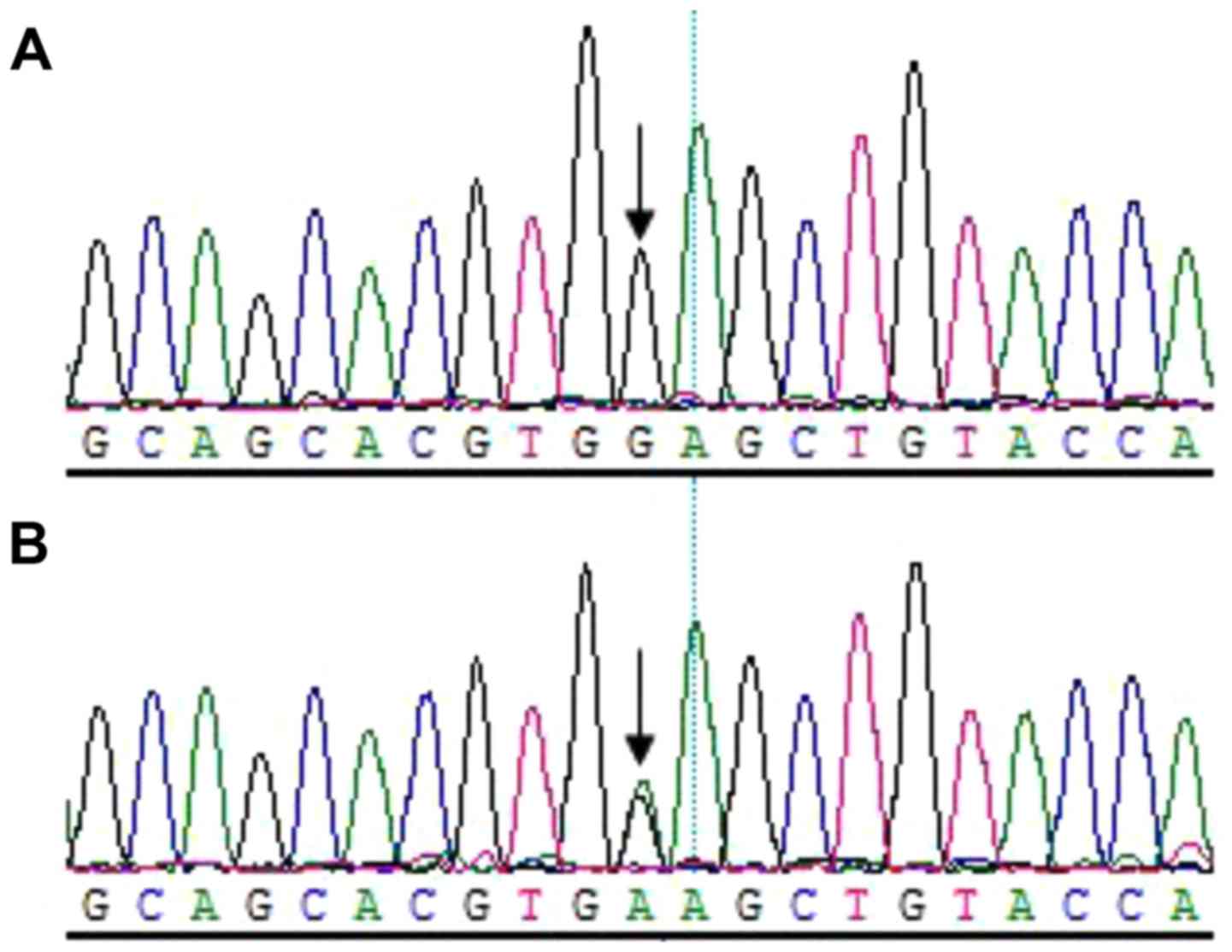
Sequencing analysis revealed that all patients
harbored the c.505G→A (p.Glu169Lys) mutation within exon 2 of
TGFβ1; however, not the healthy relatives in the family
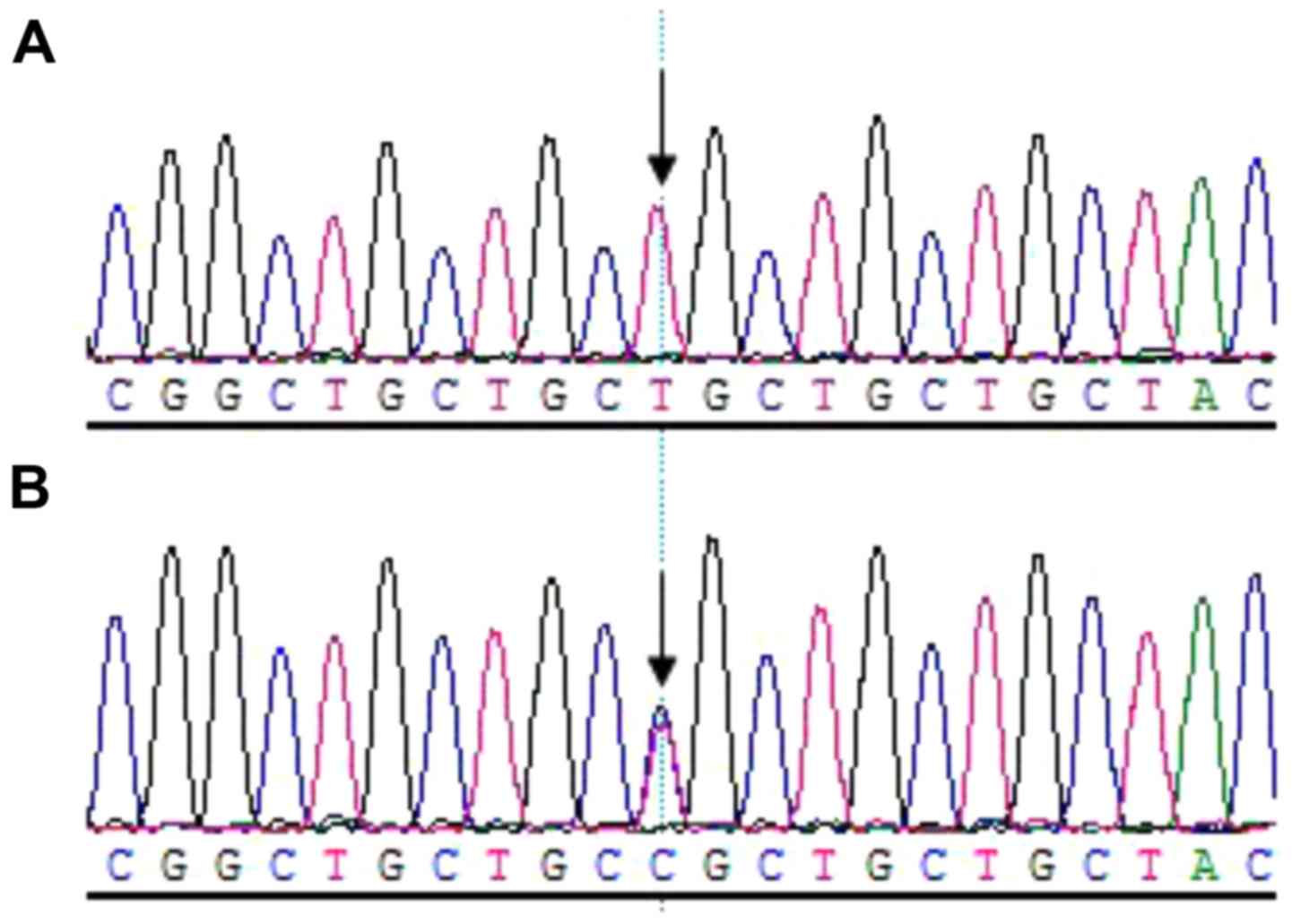
(Fig. 6). Additionally, a C→T
transition mutation at code29 (c.29C→T, p.Pro10Leu) within exon1 of
TGFβ1 was identified in one allele of the patient IV.2. The
family member III.5 (father of the patient IV.2), who was of a
normal phenotype, harbored the 29 C→T transition mutations in both
alleles (Fig. 7).
Discussion
CED is a rare bone disorder (9). The skeletal abnormalities associated
with the disease lead to a low quality of life (10,11).
However, due to the rarity and variable penetrance of CED,
diagnosis of the disease is a great challenge for clinicians
(7,8). The clinical and genetic data from the
reported CED families may guide clinicians to make the diagnosis of
novel cases.
Janssens et al (2) prepared a comprehensive review of 100
cases from 24 CED families spread worldwide and concluded that
patients with CED most commonly present pain in extremities (68%),
waddling gait (48%), easy fatigability (44%) and muscle weakness
(39%) (2). In the present study,
3/4 patients complained of pain in the extremities. Waddling gait,
easy fatigability and muscle weakness were identified for all
patients in the family. Of note, patient IV.2 appeared to be less
severely affected, not manifesting pain in extremities or the
progressive symptoms (dizziness, vision change and hearing loss),
unlike her elder relatives who were affected by CED. The present
study inferred that the differential diagnosis between the IV.2 and
other patients in the family reflects the variable penetrant nature
of CED. Therefore, clinicians must take note of young patients who
may be neglected due to incomplete penetrance of the disease.
TGFβ1 encodes an important cytokine that
serves regulatory roles in bone formation and resorption.
Therefore, abnormal activity of TGFβ1 causes bone diseases
(12). It has been previously
demonstrated that mutations in TGFβ1 are the genetic cause
of CED (5,6). To date, a series of TGFβ1
mutations associated with the pathogenesis of CED have been
identified. Among them are the L10-12 duplication, Y81H, R156C,
R218C/H and C223R/S mutation (2).
These mutations eventually lead to increased cytokine activity via
different activation mechanisms (12). Sequencing analysis revealed that
all patients in the Chinese family possessed the c.505G→A (p.
Glu169Lys) mutation within exon 2 of TGFβ1; however, not the
healthy relatives. The present study noticed that Wu et al
previously reported the E169K mutation as a genetic cause for CED,
without discussing the clinical symptoms (13). Regarding the genetic diagnosis, the
present results further confirmed that this mutation is a causative
mutation of CED. At the molecular level, the E169K mutation may be
responsible for an increase in the activity of TGFβ. However,
further mechanistic studies must be performed to confirm this
hypothesis. With the exception of the c.505G→A (p.Glu169Lys)
mutation, patient IV.2 harbored another missense mutation c.29C→T
(p.Pro10Leu) in exon 1 of TGFβ1. However, the present study
hypothesized that this transition belongs to a non-synonymous
single nucleotide polymorphism and may not be associated with the
disease, since the III.5 (father of IV.2), who had a normal
phenotype, is a homozygote of the T allele of 29C/T single
nucleotide polymorphism.
ALP is a marker of bone turnover (14). It is reported that the levels of
serum ALP were elevated in a large fraction of patients with CED
(3). The present study observed
that the levels of serum ALP in all patients in the Chinese family
were in the normal range, suggesting that serum ALP may not
necessarily reflect bone metabolism in Chinese patients with CED.
Notably, it was revealed that the level of PTH markedly increased
in the patients. It is well known that PTH/PTH-receptor signaling
serves a role in skeletal development, bone turnover and mineral
ion homeostasis (15). Previously,
Jang et al (16)
demonstrated that intermittent PTH treatment increases the number
of periosteal osteoblasts by delaying the transformation of mature
osteoblasts into lining cells on the periosteal surfaces (16). This finding and the present
observation may imply the potential role of PTH in diagnosis of
CED.
In conclusion, the present study identified a
Chinese family with CED based on the results of clinical,
biochemical and radiological examinations. The present study
reminds us that a comprehensive evaluation must be performed for
the patients prior to the final diagnosis of CED, due to its rarity
and variable phenotypes. The present data are useful to clinicians
for the diagnosis and genetic counseling of CED.
Acknowledgements
The authors would like to thank the Chinese family
for their participation in the present study. This study was
supported by grants from the Science and Technology Project of
Hunan Province (grant no. 2015JC3127) and the Health and Family
Planning Commission of Hunan Province (grant nos. B2016083 and
C2016038).
References
|
1
|
Wallace SE and Wilcox WR:
Camurati-Engelmann DiseaseGene Reviews (R). University of
Washington; Seattle (WA): 1993
|
|
2
|
Janssens K, Vanhoenacker F, Bonduelle M,
Verbruggen L, Van Maldergem L, Ralston S, Guañabens N, Migone N,
Wientroub S, Divizia MT, et al: Camurati-Engelmann disease: Review
of the clinical, radiological and molecular data of 24 families and
implications for diagnosis and treatment. J Med Genet. 43:1–11.
2006.PubMed/NCBI
|
|
3
|
Bartuseviciene A, Samuilis A and Skucas J:
Camurati-Engelmann disease: Imaging, clinical features and
differential diagnosis. Skeletal Radiol. 38:1037–1043. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Carlson ML, Beatty CW, Neff BA, Link MJ
and Driscoll CL: Skull base manifestations of Camurati-Engelmann
disease. Arch Otolaryngol Head Neck Surg. 136:566–575. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Janssens K, Gershoni-Baruch R, Guañabens
N, Migone N, Ralston S, Bonduelle M, Lissens W, Van Maldergem L,
Vanhoenacker F, Verbruggen L and Van Hul W: Mutations in the gene
encoding the latency-associated peptide of TGF-beta 1 cause
Camurati-Engelmann disease. Nat Genet. 26:273–275. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kinoshita A, Saito T, Tomita H, Makita Y,
Yoshida K, Ghadami M, Yamada K, Kondo S, Ikegawa S, Nishimura G, et
al: Domain-specific mutations in TGFB1 result in Camurati-Engelmann
disease. Nat Genet. 26:19–20. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
de Vernejoul MC: Sclerosing bone
disorders. Best Pract Res Clin Rheumatol. 22:71–83. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lucke T, Illsinger S, Das AM, Schirg E and
Hartmann H: Pitfalls in paediatric gait disturbances: Painless bone
diseases. Eur J Pediatr. 165:909–912. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cohen MM Jr: The new bone biology:
Pathologic, molecular, and clinical correlates. Am J Med Genet A.
140:2646–2706. 2006.PubMed/NCBI
|
|
10
|
Ayyavoo A, Derraik JG, Cutfield WS and
Hofman PL: Elimination of pain and improvement of exercise capacity
in Camurati-Engelmann disease with losartan. J Clin Endocrinol
Metab. 99:3978–3982. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Toumba M, Neocleous V, Shammas C,
Anastasiadou V, Allgrove J, Phylactou LA and Skordis N: A family
with Camurati-Engelman disease: The role of the missense p. R218C
mutation in TGFbeta1 in bones and endocrine glands. J Pediatr
Endocrinol Metab. 26:1189–1195. 2013.PubMed/NCBI
|
|
12
|
Wang C, Zhang BH, Liu YJ, Hu YQ, He JW and
Zhang ZL: Transforming growth factor-β1 gene mutations and
phenotypes in pediatric patients with Camurati-Engelmann disease.
Mol Med Rep. 7:1695–1699. 2013.PubMed/NCBI
|
|
13
|
Wu S, Liang S, Yan Y, Wang Y, Li F, Deng
Y, Huang W, Yuan W, Luo N, Zhu C, et al: A novel mutation of TGF
beta1 in a Chinese family with Camurati-Engelmann disease. Bone.
40:1630–1634. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hernàndez MV, Peris P, Guañabens N,
Alvarez L, Monegal A, Pons F, Ponce A and Muñoz-Gómez J:
Biochemical markers of bone turnover in Camurati-Engelmann disease:
A report on four cases in one family. Calcif Tissue Int. 61:48–51.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cheloha RW, Gellman SH, Vilardaga JP and
Gardella TJ: PTH receptor-1 signalling-mechanistic insights and
therapeutic prospects. Nat Rev Endocrinol. 11:712–724. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jang MG, Lee JY, Yang JY, Park H, Kim JH,
Kim JE, Shin CS, Kim SY and Kim SW: Intermittent PTH treatment can
delay the transformation of mature osteoblasts into lining cells on
the periosteal surfaces. J Bone Miner Metab. 34:532–539.
2015.PubMed/NCBI
|