Introduction
Triphalangeal thumb-polysyndactyly syndrome (TPT-PS;
OMIM #174500), also called pre-axial polydactyly type II (PPD2;
OMIM #174500), is a well-defined autosomal dominant disorder with
complete penetrance and variable expression. The phenotype consists
of opposable triphalangeal thumbs, duplication of distal thumb
phalanx, pre-axial polydactyly, and duplication of the big toes
(hallux).
Using linkage analysis, a malformation linked,
highly polymorphic locus was identified within a large Dutch family
with TPT-PS and, for the first time, the disease locus was located
at the 7q36 region (1). Variable
expression of TPT-PS was demonstrated by the asymmetrical limb
deformities of affected individuals and the differences observed
between monozygotic twins in that family.
The Sonic hedgehog (SHH) protein secreted by the
mesenchymal cells is restricted to a region termed the zone of
polarizing activity (ZPA) in the limb bud as a key regulator in
defining the anterior-posterior axis and numbers of digits in early
limb development and morphogenesis (2,3). In
2002, a translocation breakpoint was first identified in a
pre-axial polydactyly patient (4).
In 2003, it was reported that point mutations segregated with
polydactyly in three unrelated families with PPD2 and in the
Hemimelic extra toes mouse mutant. A further study identified a
cis-regulator, designated ZPA regulatory sequence (ZRS),
driving normal SHH expression in ZPA, which lies at 1 Mb upstream
of the target gene SHH within intron 5 of the Limb region 1
homolog (LMBR1; OMIM #605522) (5). The ZRS is highly conserved from
mammals to fish and can drive the expression of a reporter gene in
ZPA (5,6). Subsequently, several different point
mutations that are clustered within a highly conserved region of
ZRS in humans have been reported to be associated with TPT-PS
(7–9).
In 2008, for the first time in humans, a 588,819 bp
duplication of 7q36 comprising ZRS was reported in a large family
with TPT-PS (10). This previous
study demonstrated that a duplication of ZRS results in a similar
phenotype, that is, TPT-PS as single nucleotide mutations within
ZRS. In the same year, six duplications of at least 131, 291, 158,
246, 235 and 398 kb were identified in six families with TPT-PS
(11). These duplications shared a
32,757 bp common overlapping segment containing the ZRS
enhancer.
A previous study (12) suggested that copy number variations
(CNVs) can be associated with syndromic congenital heart disease
(CHD), including limb anomalies. The present study reported the
identification of a novel 0.29 Mb duplication in 7q36.3
encompassing ZRS in LMBR1 in a 1-year-old female patient
with the TPT-PS phenotype and CHD associated with a de novo
22q11.21 deletion.
Materials and methods
Ethical approval and patient
consent
The present study was approved by the Review Board
of The Second Xiangya Hospital of the Central South University
(Changsha, China). All individuals provided informed written
consent. Blood samples were collected following informed written
consent from the patient's parent on behalf of the child
enrolled.
Clinical presentation
The present study investigated a four-generation
family from Central-South China (Hunan) with a phenotype comprising
triphalangeal thumbs and pre-axial polysyndactyly leading to the
diagnosis of TPT-PS, and the phenotype varied among affected
individuals. The proband, a 1-year-old female patient, was the
second child of non-consanguineous parents. Central cyanosis,
cardiac murmur and a malformation of both thumbs were observed at
birth.
At 1 year of age, the patient was referred to the
in-patient department of The Second Xiangya Hospital of the Central
South University (Changsha, Hunan, China) for surgical treatment of
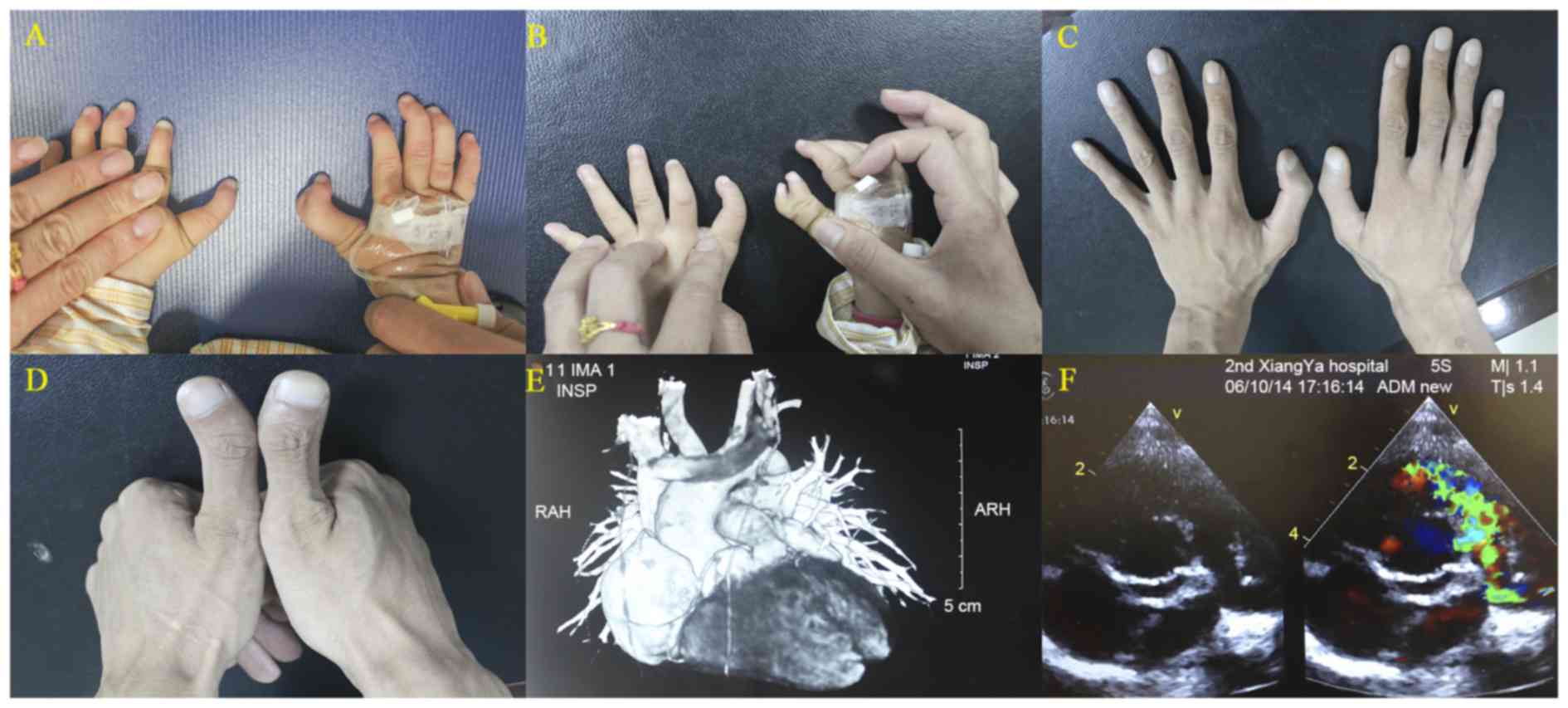
cardiac defects. Tetralogy of Fallot (TOF) was diagnosed by
two-dimensional color Doppler echocardiography. Physical
examination highlighted the limb anomalies of the proband, which
comprised triphalangeal thumbs of both hands and polysyndactyly in
the right thumb, and so typical TPT-PS was diagnosed (Fig. 1). Next, the patient's father was
also ascertained to be expressing triphalangeal thumbs bilaterally
but without polysyndactyly, and CHD was excluded using color
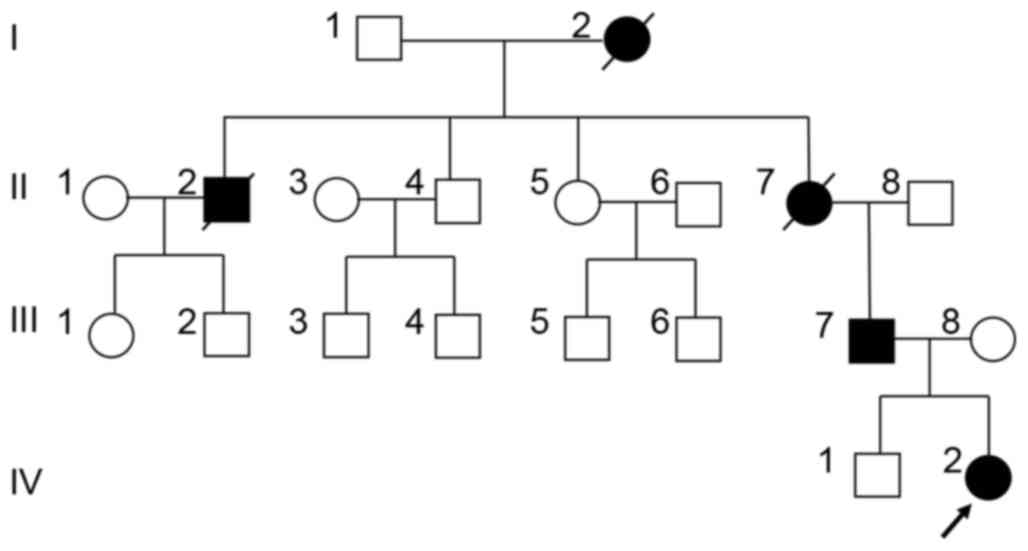
Doppler echocardiography. A pedigree analysis revealed that the
proband's grandmother, one of the grandmother's brothers and the
grandmother's mother manifested similar thumb phenotypes, but had
all died a number of years previously. This indicated that the
phenotype was transmitted in an autosomal dominant manner
throughout four generations in the family (Fig. 2). The pedigree had no history of
CHD, with the exception of the female patient with TOF.
Cytogenetic analysis
Chromosome analysis was performed on the peripheral
blood samples of the patient and the patient's parents by
conventional G-Banded techniques (550 bands resolution). A 2 ml
sample of peripheral blood was collected from each individual. All
samples were subjected to lymphocyte culture according to standard
cytogenetic protocol.
DNA extraction
Genomic DNA was prepared from peripheral blood using
a DNeasy Blood and Tissue kit (Qiagen, Inc., Valencia, CA, USA) on
the QIAcube automated DNA extraction robot (Qiagen GmbH, Hilden,
Germany) following the manufacturer's protocol.
Sequence analysis
The entire coding regions, including the flanking
intronic sequences of LMBR1 (RefSeq: NM_022458) and TBX1 (RefSeq:
NM_080647) were amplified with polymerase chain reaction (PCR;
primer sequences are presented in Table I and the supplier was Sangon
Biotech Co., Ltd., Shanghai, China). Sequences of the PCR products
were determined using the ABI 3100 Genetic Analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA), as
previously described (13).
 | Table I.Primer sequences used to amplify ZRS
(on LMBR1) and TBX1. |
Table I.
Primer sequences used to amplify ZRS
(on LMBR1) and TBX1.
| Gene | Exon | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| LMBR1 | ZRS-1 |
TTTCAAATGCTCACTTTACATGG |
TTTTATGACCAGATGACTTTTTCC |
|
| ZRS-2 |
AGGCTGGACTTCCTACTCACTCT |
GAATAAAAATGTCAGGAGGAAAAA |
| TBX1 | 2 |
AGCCAGTGCGTTCAGCATCGCCTC |
AAGAGCTGCCTCCACCTACTTTG |
|
| 3-1 |
TGATCTCCGCCGTGTCCAGC |
GGCCACCTTCGCGTTCTTCTT |
|
| 3-2 |
GGTGAAGAAGAACGCGAAGGTGG |
GACGGCGAACAGCGAAGGAG |
|
| 4 |
TGCCTTCCACCAGCTAGG |
AGACGACCCTTGGAGTTGG |
|
| 5 |
CTCTGGGTTCACCTCCACAT |
CAGGCCTCTTAGGGACAGG |
|
| 6 |
CTCCCACCCCAGATCCTC |
AATCCGCTCAGGTCCTCC |
|
| 7 |
TTGGTGCGCTTCTCCTAACACTC |
AAGGCGCTCATGAGCGGCAGTG |
|
| 8 |
GGGACTGTGACCCTGAGGACTG |
GGGTAGAGCGCGCACAGG |
|
| 9A-1 |
TCAGACACTGGACATTTGTGC |
TCAGCATTCAACAAAGACGG |
|
| 9A-2 |
CAGAGTGCCAACCCTTCAAT |
ACTGGGAGTGTGACTCTATGGA |
|
| 9B |
AACACTTTGACCTTCCTCCACCC |
TGGAGGATTCGCTTCCATCACAG |
|
| 9C-1 |
TCGCATGGGGCGTCGGAGCT |
CCCGAGATAGTGGTCGTAGGC |
|
| 9C-2 |
GGTGCTAAGCCCCTCGCTG |
GCCGACGAGTACATGTTGGC |
|
| 9C-3 |
ACGGCTACCACCCGCACG |
TATTCCTTGCTTGCCCTTGG |
Single nucleotide polymorphism (SNP)
array analysis
Genomic DNA samples were adjusted to a final
concentration of 50 ng/ml. The Human Omni1-Quad Chip (Illumina,
Inc., San Diego, CA, USA) and the Illumina BeadScan genotyping
system (Beadstation Scanner) were employed to obtain the signal
intensities of SNP probes. The Genome Studio software (version
2011; Illumina, Inc.) was used to analyze the genotypes [human
genome build 37 (Hg19)] and to evaluate the experimental quality.
The call rates of the samples were >99.0%.
Results
The karyotypes of the patient and the patient's
parents were normal. Sequence analysis did not identify any
missense or nonsense mutation in LMBR1 and TBX1. To
explore the presence of genomic imbalances, an SNP-array system was
employed to analyze the whole genome for CNVs (Human Omni1-Quad
Chip, Beadstation Scanner and GenomeStudio V2011 software). A total
of 343 CNVs were identified. By comparing these CNVs with the
Database of Genomic Variants (http://dgv.tcag.ca), the present study identified a
novel 0.29 Mb duplication of chromosome 7q36.3 (Chr7:
156,484,201–156,772,643), which contains two genes, LMBR1
and NOM1 (Fig. 3). The
patient's TPT-PS affected father also harbored the same
duplication, however, the patient's TPT-PS unaffected mother did
not.
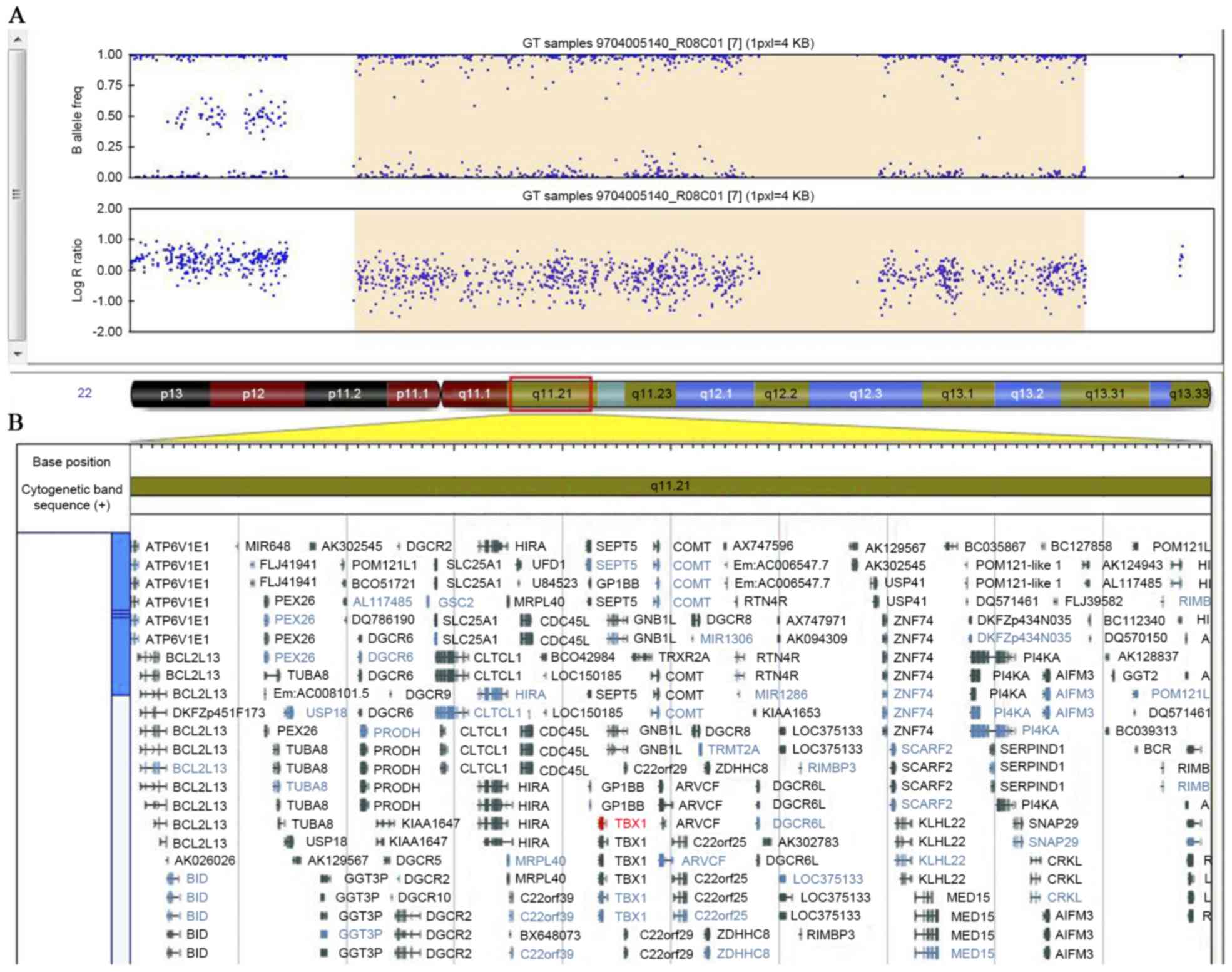
In addition, the present study also identified a
de novo 2.6 Mb deletion of chromosome 22q11.21 (chr22:
18,874,965–21,464,479) in the proband. This 2.6 Mb deletion region
includes TBX1, HIRA, RTN4R and numerous other
genes (Fig. 4). However, the
proband's parents did not harbor the 22q11.21 deletion.
Discussion
The present study reported a complication that
multiple pathogenic CNVs co-exist in one patient, and consequently
resulted in congenital multi-system diseases, complicating limb
anomalies with congenital heart defects.
The duplicated region of chromosome 7q36.3 in the
patient reported in the present study spans 288,443 bp containing
the genes LMBR1 and, in part, NOM1. To the best of
our knowledge, this is a novel 7q36.3 duplication. The duplication
was identified to co-segregate with the TPT-PS phenotype in the
patient and the patient's father, but was not present in the family
members unaffected by TPT-PS. This result notably suggested that
the duplication involving ZRS is the disease-causing mutation
underlying human TPT-PS.
Intron 5 of LMBR1 contains a
cis-acting regulator of limb-specific SHH, which is
designated as ZRS (2,5). The role of ZRS in limb development
has been interpreted as an enhancer driving SHH expression in the
posterior limb bud, as well as a repressor that silences the
anterior expression. The direct link between ZRS and the PPD
phenotypes was established by the identification of a number of the
different point mutations within ZRS, including in polydactylous
cats, in mouse models of PPD, and in human PPD families (5–7,14).
Duplication has been shown to be a major type of CNV in numerous
human genetic diseases (15). One
possible explanation is a gain of function mechanism leading to an
augmented SHH expression in the limb bud. The pathogenicity of the
duplication involving ZRS may be further supported by a previous
study (16) in the Sasquatch
mutant mouse whose phenotype is an isolated pre-axial
polydactylous. This mutant was generated by the transgenic
insertion of a reporter gene in intron 5 of Lmbr1 and has
been shown to have a 24 kb intronic duplication containing the
mouse ZRS (5). In 2008, Klopocki
et al (10) first reported
the identification of a genomic duplication containing ZRS in a
large German family with TPT-PS. In the same year, Sun et al
(11) identified six duplications
in six families with TPT-PS. These independent findings and the
results of the present study consistently implied that the
duplication of ZRS long range enhancer is the molecular
pathogenesis of TPT-PS. It is hypothesized that disruption of
cis-regulator via ZRS duplication results in the
dysregulation of SHH, leading to TPT-PS phenotypes.
The deletion of chromosome 22q11.21 in the patient
reported in the present study spans 2,589,515 bp, containing
TBX1, HIRA, RTN4R and numerous other genes.
Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome; OMIM
#188400) is one of the most common genetic syndromes, with a
prevalence of 1:4,000 to 1:6,000 (17,18).
At least 30 genes have been mapped to the deleted region, among
them TBX1, a major genetic determinant of the 22q11.2
deletion syndrome (19). The
phenotypic spectrum is varied, including CHD, velopharyngeal
insufficiency and cleft palate facial anomalies, immune disorders,
and hypocalcemia secondary to hypoparathyroidism (20). The most common cardiac defects are
conotruncal anomalies, including TOF, TOF with pulmonary atresia,
truncus arteriosus and interrupted aortic arch (21). In the present study, the patient
was diagnosed as TOF, but presented neither obvious facial
anomalies nor immune deficiencies. Her parents did not harbor the
22q11.21 deletion and did not have CHD. It is inferred that it was
the patient's deletion of 22q11.21 that caused the CHD and TOF.
This is the first report, to the best of our knowledge, on a
co-existence of 7q36.3 duplication with 22q11.21 deletion in a
syndromic CHD.
In conclusion, the present study described a
duplication of 7q36.3 containing the ZRS long range enhancer of SHH
in a family with distinctive phenotypic features of TPT-PS.
Additionally, a de novo 22q11.21 deletion containing
TBX1 was detected in the proband who is the unique member of
the family with TOF, a type of CHD. The findings of the present
study indicated that 7q36.3 duplication encompassing ZRS region is
the underlying genetic cause of TPT-PS, and in addition,
corroborate that 22q11.21 deletion is a genetic cause of CHD.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (no. 8140020813 to Cheng Luo).
The authors would like to thank the patient and her parents for
participating in this study. The authors are also grateful to the
State Key Laboratory of Medical Genetics of China, in particular to
Mr Qian Pan and Miss Yi-Qiao Hu, for technical support.
References
|
1
|
Tsukurov O, Boehmer A, Flynn J, Nicolai
JP, Hamel BC, Traill S, Zaleske D, Mankin HJ, Yeon H and Ho C: A
complex bilateral polysyndactyly disease locus maps to chromosome
7q36. Nat Genet. 6:282–286. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hill RE: How to make a zone of polarizing
activity: Insights into limb development via the abnormality
preaxial polydactyly. Dev Growth Differ. 49:439–448. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Radhakrishna U, Blouin JL, Solanki JV,
Dhoriani GM and Antonarakis SE: An autosomal dominant triphalangeal
thumb: Polysyndactyly syndrome with variable expression in a large
Indian family maps to 7q36. Am J Med Genet. 66:209–215. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lettice LA, Horikoshi T, Heaney SJ, van
Baren MJ, van der Linde HC, Breedveld GJ, Joosse M, Akarsu N,
Oostra BA, Endo N, et al: Disruption of a long-range cis-acting
regulator for Shh causes preaxial polydactyly. Proc Natl Acad Sci
USA. 99:7548–7553. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lettice LA, Heaney SJ, Purdie LA, Li L, De
Beer P, Oostra BA, Goode D, Elgar G, Hill RE and de Graaff E: A
long-range Shh enhancer regulates expression in the developing limb
and fin and is associated with preaxial polydactyly. Hum Mol Genet.
12:1725–1735. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sagai T, Masuya H, Tamura M, Shimizu K,
Yada Y, Wakana S, Gondo Y, Noda T and Shiroishi T: Phylogenetic
conservation of a limb-specific, cis-acting regulator of Sonic
hedgehog (Shh). Mamm Genome. 15:23–34. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gurnett CA, Bowcock AM, Dietz FR,
Morcuende JA, Murray JC and Dobbs MB: Two novel point mutations in
the long-range SHH enhancer in three families with triphalangeal
thumb and preaxial polydactyly. Am J Med Genet A. 143A:27–32. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang ZQ, Tian SH, Shi YZ, Zhou PT, Wang
ZY, Shu RZ, Hu L and Kong X: A single C to T transition in intron 5
of LMBR1 gene is associated with triphalangeal thumb-polysyndactyly
syndrome in a Chinese family. Biochem Biophys Res Commun.
355:312–317. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Al-Qattan MM, Al Abdulkareem I, Al Haidan
Y and Al Balwi M: A novel mutation in the SHH long-range regulator
(ZRS) is associated with preaxial polydactyly, triphalangeal thumb,
and severe radial ray deficiency. Am J Med Genet A. 158A:2610–2615.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Klopocki E, Ott CE, Benatar N, Ullmann R,
Mundlos S and Lehmann K: A microduplication of the long range SHH
limb regulator (ZRS) is associated with triphalangeal
thumb-polysyndactyly syndrome. J Med Genet. 45:370–375. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sun M, Ma F, Zeng X, Liu Q, Zhao XL, Wu
FX, Wu GP, Zhang ZF, Gu B, Zhao YF, et al: Triphalangeal
thumb-polysyndactyly syndrome and syndactyly type IV are caused by
genomic duplications involving the long range, limb-specific SHH
enhancer. J Med Genet. 45:589–595. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Luo C, Yang YF, Yin BL, Chen JL, Huang C,
Zhang WZ, Wang J, Zhang H, Yang JF and Tan ZP: Microduplication of
3p25.2 encompassing RAF1 associated with congenital heart disease
suggestive of Noonan syndrome. Am J Med Genet A. 158A:1918–1923.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tan ZP, Huang C, Xu ZB, Yang JF and Yang
YF: Novel ZFPM2/FOG2 variants in patients with double outlet right
ventricle. Clin Genet. 82:466–471. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Masuya H, Sezutsu H, Sakuraba Y, Sagai T,
Hosoya M, Kaneda H, Miura I, Kobayashi K, Sumiyama K, Shimizu A, et
al: A series of ENU-induced single-base substitutions in a
long-range cis-element altering Sonic hedgehog expression in the
developing mouse limb bud. Genomics. 89:207–214. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Beckmann JS, Estivill X and Antonarakis
SE: Copy number variants and genetic traits: Closer to the
resolution of phenotypic to genotypic variability. Nat Rev Genet.
8:639–646. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sharpe J, Lettice L, Hecksher-Sorensen J,
Fox M, Hill R and Krumlauf R: Identification of sonic hedgehog as a
candidate gene responsible for the polydactylous mouse mutant
Sasquatch. Curr Biol. 9:97–100. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Goodship J, Cross I, LiLing J and Wren C:
A population study of chromosome 22q11 deletions in infancy. Arch
Dis Child. 79:348–351. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Botto LD, May K, Fernhoff PM, Correa A,
Coleman K, Rasmussen SA, Merritt RK, O'Leary LA, Wong LY, Elixson
EM, et al: A population-based study of the 22q11.2 deletion:
Phenotype, incidence, and contribution to major birth defects in
the population. Pediatrics. 112:101–107. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yagi H, Furutani Y, Hamada H, Sasaki T,
Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, et
al: Role of TBX1 in human del22q11.2 syndrome. Lancet.
362:1366–1373. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shprintzen RJ: Velo-cardio-facial
syndrome: 30 Years of study. Dev Disabil Res Rev. 14:3–10. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Momma K: Cardiovascular anomalies
associated with chromosome 22q11.2 deletion syndrome. Am J Cardiol.
105:1617–1624. 2010. View Article : Google Scholar : PubMed/NCBI
|