Introduction
8p11 myeloproliferative syndrome (EMS) is a rare,
aggressive disease, which progresses to acute leukemia within 1–2
years following diagnosis (1). It
is caused by the constitutive activation of the fibroblast growth
factor receptor 1 (FGFR1) gene by fusion to various partner genes.
To date, 15 FGFR1 partner genes have been identified, including
BCR (22q11), NUP98 (11p15), HERV-K (19q13),
MYO18A (17q23), ZNF198 (13q12), CPSF6 (12q15),
FGFR1OP2 (12p11), CEP110 (9q33), TIFI (7q34),
CUX1 (7q22), FOP (6q27), LRRFIP1 (2q37),
TPR (1q25) (2),
RANBP2/NUP358 (2q12) (3) and SQSTM1 (5q35) (4).
The TPR-FGFR1 arrangement was first
identified in our previous study (2) in a patient, who presented with
myeloproliferative neoplasm-like symptoms. In this case, exon 23 of
the TPR gene was fused to exon 13 of FGFR1. Another
case of TPR-FGFR1 rearrangement, with a novel breakpoint of
exon 22 of the TPR gene, has also been reported (5). However, the biological function of
this fusion remains to be elucidated.
The constitutive activation of FGFR1 kinase is the
primary cause of EMS, which suggests the potential of FGFR1 kinase
as a promising target for the treatment of EMS. Previous studies
have shown that SU5402, PD173074 and PKC412 potently inhibit the
proliferation of ZNF198-FGFR1-transformed Baf3 cells (6). However, the results did not translate
into clinical benefits, and novel FGFR1-targeted compounds are
required for EMS therapy (7).
TKI258, ponatinib and AZD4547 are three clinically
evaluated TKIs, which target FGFR1 with enzyme half maximal
inhibitory concentration (IC50) values <10 nmol/l
(6). Previous studies have also
indicated that TKI258, ponatinib and AZD4547 are active against
certain EMS-associated fusion genes (8–11).
In the present study, the transforming activity of
TPR-FGFR1 was analyzed, and the effects of TKI258,
ponatinib, and AZD4547 on the fusion gene were compared in
vitro to identify the drug with the most potential for the
treatment of EMS.
Materials and methods
Constructs
The TPR-FGFR1/pEZ-M03 plasmid was generated using
Gateway cloning technology. Briefly, TPR1-23 was amplified from the
CCS-Z8318-M03 plasmid (FulenGen Co., Ltd., Guangzhou, China) using
the following primers: Forward
5′-GGAAGTTCGAACCATGGCGGCGGTGTTGCAGCAAGT-3′ and reverse
5′-CACTGGAGTCAGCAGACACCTGTTGTTCCATGCTCTCTATGGC-3′. DNA was first
denatured for 10 min at 94°C, and then amplified using 25 cycles of
30 sec at 94°C, 30 sec at 58°C and 2 min at 72°C. The FGFR1 moiety
was liberated from the EX-Z0528-M03 plasmid (FulenGen Co., Ltd.)
using the following primers: Forward
5′-GCCATAGAGAGCATGGAACAACAGGTGTCTGCTGACTCCAGTG-3′ and reverse
5′-TGCGGCCGCACTCGAGGTAGCGGCGTTTGAGTCCGCCATTGG-3′. The TPR and FGFR1
fragments were ligated into the pDONR™ vector (FulenGen Co., Ltd.)
using a Fast-Fusion™ kit (FulenGen Co., Ltd.). The fusion gene was
then transferred into pEZ-M03/GFP via the Gateway LR cloning
reaction (EZShuttle™ LR Recombination Cloning System, Genecopoeia,
Rockville, MD, USA).
The full-length TPR-FGFR1 was amplified from
the TPR-FGFR1/pEZ-M03 plasmid using the following primers: Forward
5′-CTTAGAATTCGCCACCATGGCGGCGGTGTTGCAGCAAGTCCTGGAGCGCACGGA-3′ and
reverse 5′-CTTACTCGAGCTAGCGGCGTTTGAGTCCGCCATTGGCAAGCTGGGCTGGGTG-3′.
The target DNA was amplified using 25 cycles of 30 sec at 94°C, 30
sec at 58°C and 2.5 min at 72°C. This was then cloned into the
EcoRI/XhoI site of the pMSCV (ClontechLaboratories,
Inc., Mountainview, CA, USA).
The successful construction of TPR-FGFR1/pEZ-M03 and
TPR-FGFR1/pMSCV were validated using gene sequencing.
Cell lines
All cell lines were obtained from the China
Infrastructure of Cell Line Resources (Beijing, China). Baf3 cells
were cultured in RPMI 1640 supplemented with 10% fetal calf serum
containing interleukin (IL) 3 (10 ng/ml; eBioscience, Inc., Vienna,
Austria). COS-1 and 293T cells were cultured in Dulbecco's modified
Eagle's medium containing 10% newborn calf serum (Zhejiang
Evergreen Biologicals, Zhejiang, China).
Subcellular localization of
TPR-FGFR1
COS-1 cells (1×105/ml) were cultured (5%
CO2, 37°C) in a six-well microplate with a
polylysine-coated coverslip. After 24 h, the cells were transfected
with TPR-FGFR1/pEZ-M03 using Thermo Scientific TurboFect
transfection reagent (Thermo Fisher Scientific, Inc., Rockford, IL,
USA) according to the manufacturer's protocol. Subsequently, the
protein localization (green fluorescence) was detected using
fluorescence microscopy.
Establishment of TPR-FGFR1-expressing
Baf3 cells
Retroviral stock was generated by co-transfection of
293T cells with TPR-FGFR1/pMSCV or empty vector and two packaging
constructs (VSVG and MD2) provided by Professor Quan Zhao (Nanjing
University, Nanjing, China). At 48 h post-transfection, the
supernatant was collected and stored at −70°C. For the
transfection, the virus-containing supernatant was added to the
Baf3 cells (5×105/ml) together with 10 mg/ml polybrene, and
cultured at 37°C for 72 h. The cells were then selected by the
addition of 1 mg/ml G418. The medium was replaced every 4 days.
After 5 weeks, the resistant cells were collected for use in the
subsequent experiments.
Fusion gene characterization
RNA was extracted from cells using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), and 1 µg RNA was
reverse transcribed into cDNA with PrimeScript™ RT Master Mix
(Takara Bio, Inc., Otsu, Japan) according to the manufacturer's
protocol. Polymerase chain reaction (PCR) analysis was performed
using a BIO-RAD T100 Thermal Cycler (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The primer combinations used to detect the
fusion gene were as follows: Sense, 5′-GCCACATTGAAACAGCACCTC-3′ and
antisense 5′-AGCCGTGATGGCCGAAC-3′. The target DNA was amplified
with Takara PCR Amplification Kit (Takara Bio, Inc.) using 30
cycles of 30 sec at 94°C, 30 sec at 60°C, and 40 sec at 72°C.
Proliferation assays
The cells were seeded into a 96-well plate at
1×104/ml (100 µl/well) with or without IL3 (10 ng/ml) at 37°C. The
growth curve was constructed by counting the number of viable
cells, which excluded trypan blue, each day with microscope
(Olympus CX41). The selected cells were then seeded into a 96-well
plate at 1×105/ml (100 µl/well) and treated with different
concentrations of the TKIs (Selleck Chemicals, Shanghai, China),
TKI258, ponatinib and AZD4547, ranging between 1 and 1,000 nmol/l
with half-log increments at 37°C. Cells treated with DMSO were used
as a control. After 48 h, 20 ml of 5 mg/ml
methylthiazolyldiphenyl-tetrazolium bromide was added to each well,
and cultured at 37°C for 4 h. The supernatant was removed carefully
following centrifugation at 1,600 × g, room temperature, for 30
min. Subsequently, 100 µl DMSO was added and absorbance was
detected at 490 nm (Bio-Rad iMark Microplate Reader; Bio-Rad
Laboratories, Inc.).
Apoptosis assay
Cells were treated with the different concentrations
of the TKIs for 24 h. Subsequently, apoptosis was detected using an
Annexin V/PI apoptosis kit (7Sea Biotechnology, Shanghai, China)
according to the manufacturer's protocol. The stained cells were
then detected using the FACS Calibur system (Miltenyi Biotec GmbH,
Cologne, Germany).
Western blot analysis
The cells were serum starved overnight and treated
with the different concentrations of the TKIs for 90 min. The cells
were lysed on ice with RIPA cell lysis buffer containing 1 mmol/l
PMSF (Beyotime Institute of Biotechnology, Shanghai, China). The
lysed cells were harvested, transferred into new tubes, and
centrifuged for 10 min at 12,000 × g at 4°C, following which the
supernatants were collected. The protein concentrations were
determined using a BCA kit (Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China). A total of 30 mg protein per
group was electrophoresed on a 30% sodium dodecyl
sulfate-polyacrylamide electrophoresis gel and transferred onto a
PVDF membrane. Following three washes with TBST, the membrane was
blocked by 5% BSA (Sigma-Aldrich; Merck Millipore, Darmstadt,
Germany) for 2 h at room temperature. The primary antibodies,
rabbit anti-FGFR1 (cat. no. sc-121; 1:100; Santa Cruz Biotechnology
Inc., Dallas, TX, USA), rabbit anti-phospho-FGFR1 (cat. no. 3461;
1:1,000; Cell Signaling Technology, Inc. Beverly, MA, USA), rabbit
anti-phospho-signaling transducer and activator of transcription 5
(STAT5; cat. no. 4322; 1:1,000; Cell Signaling Technology, Inc.),
rabbit anti-phospho-phospholipase γ (PLCγ; cat. no. 8713; 1:1,000;
Cell Signaling Technology, Inc.), rabbit anti-phospho-extracellular
signal-regulated kinase (ERK; cat. no. 4695; 1:1,000; Cell
Signaling Technology, Inc.), rabbit anti-phospho-Akt (cat. no.
2965; 1:1,000; Cell Signaling Technology, Inc.) and rabbit
anti-β-actin (cat. no. AA128; 1:1,000; Beyotime Institute of
Biotechnology), were used for the incubation overnight at 4°C.
Following this, the membrane was washed with TBST for 10 min at
room temperature three times. Following 2 h of incubation with
horseradish peroxidase-conjugated anti-rabbit IgG antibody (cat.
no. A0208, 1:1,000, Beyotime Institute of Biotechnology) at room
temperature, the membrane was washed three times with TBST and
detected with enhanced chemiluminescence (GE Healthcare Life
Sciences, Little Chalfont, UK).
Statistical analysis
The results are expressed as the mean ± standard
error of the mean. Student's t-test was used to compare data with
SPSS software version 19.0 (IBM SPSS, Armonk, NY, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
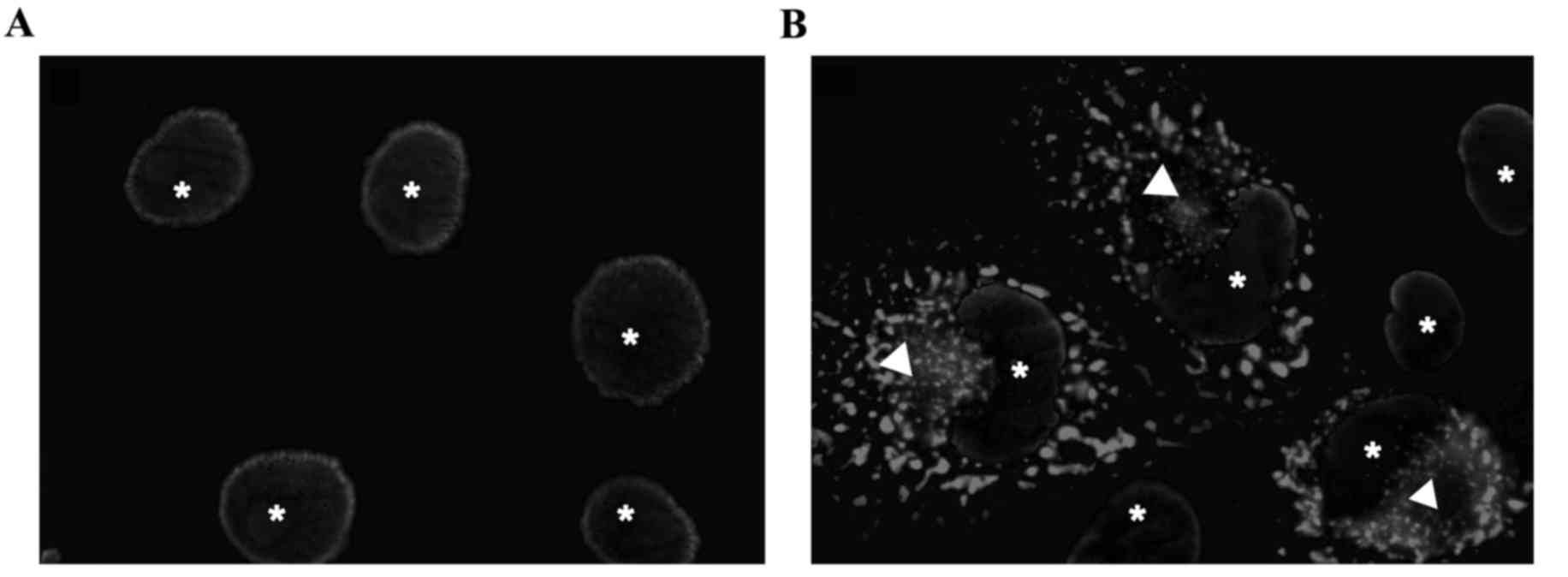
TPR-FGFR1 is localized in the
cytoplasm
In order to investigate the subcellular localization
of the fusion gene, COS-1 cells were transiently transfected with
TPR-FGFR1/pEZ-M03. GFP was conjugated to the carboxyl terminus of
the fusion gene. As shown in Fig.
1, TPR-FGFR1 was exclusively localized in the cytoplasm.
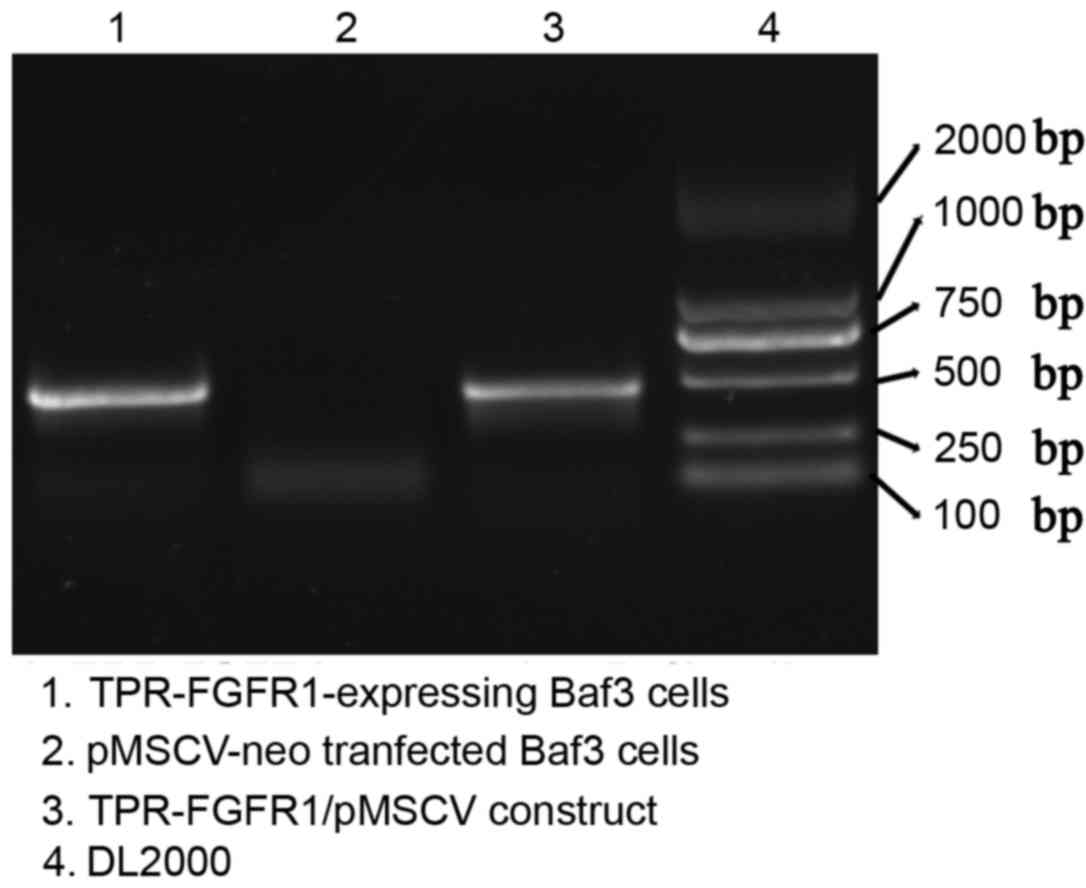
TPR-FGFR1 is expressed in
TPR-FGFR1-transfected Baf3 cells
The selected TPR-FGFR1-transfected Baf3 cells were
analyzed for the expression of the fusion gene using reverse
transcription (RT)-PCR analysis. Empty vector
(pMSCVneo)-transfected cells were used as a negative control. The
PCR product of the TRP-FGFR1/pMSCV construct, which was confirmed
using gene sequencing, was also electrophoresed as a positive
control. Data showed that TPR-FGFR1 was expressed in the selected
Baf3 cells (Fig. 2). These cells
were referred to as TPR-FGFR1-expressing Baf3 cells in the
subsequent experiment.
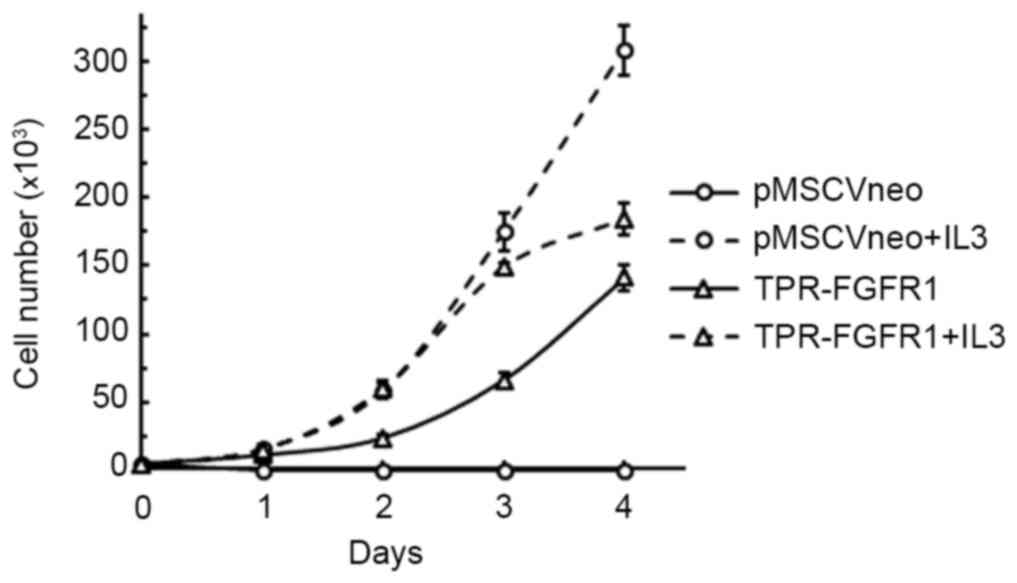
TPR-FGFR1 transforms Baf3 cells into
IL3-independent cells
The growth dependence of IL3 was evaluated to
investigate the transforming activity of TPR-FGFR1. As shown in
Fig. 3, all the cells transfected
with the empty vector (pMSCVneo) died within 2 days, whereas the
TPR-FGFR1-expressing Baf3 cells were found to grow in an
IL3-independent manner. The growth rate of the TPR-FGFR1-expressing
Baf3 cells was marginally lower, compared with that in the
pMSCV-neo transfected cells with IL3.
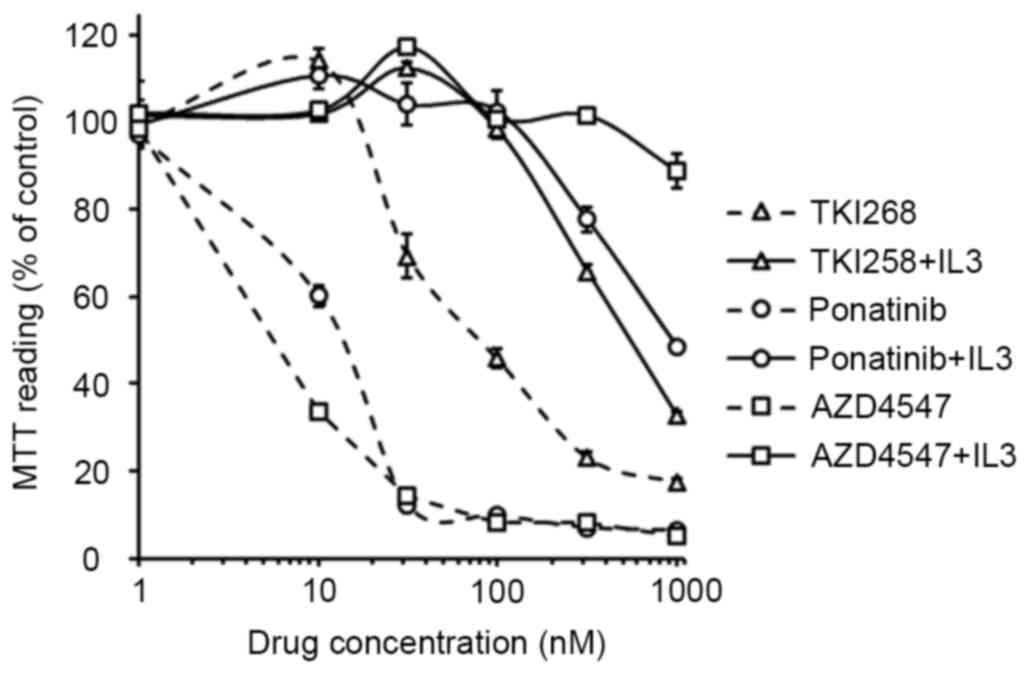
TKI258, ponatinib and AZD4547 inhibit
the proliferation of TPR-FGFR1-expressing Baf3 cells
Previous studies have investigated the activities of
TKI258, ponatinib and AZD4547 against ZNF198-FGFR1, BCR-FGFR1,
CUX1-FGFR1 and FGFR1OP2-FGFR1 (8–11).
However, the effects of these agents on TPR-FGFR1 remain to be
elucidated. In the present study, their effects on the
proliferation of TPR-FGFR1-expressing Baf3 cells were examined. The
data showed that the treatment of TPR-FGFR1-expressing Baf3 cells
with TKI258, ponatinib and AZD4547 significantly inhibited cell
growth, with IC50 values of 112.374, 13.506 and 8.503
nmol/l, respectively. TKI258 treatment did not affect cell
proliferation at the concentration of 10 nmol/l. By contrast,
incubation with 10 nmol/l ponatinib resulted in significant cell
growth inhibition (P<0.01), and treatment with AZD4547 at the
same concentration further inhibited the proliferation (P<0.01).
The addition of IL3 resulted in IC50 values of >500
nmol/l (Fig. 4), suggesting that
the effect of the three drugs were specifically mediated by FGFR1
signaling (9).
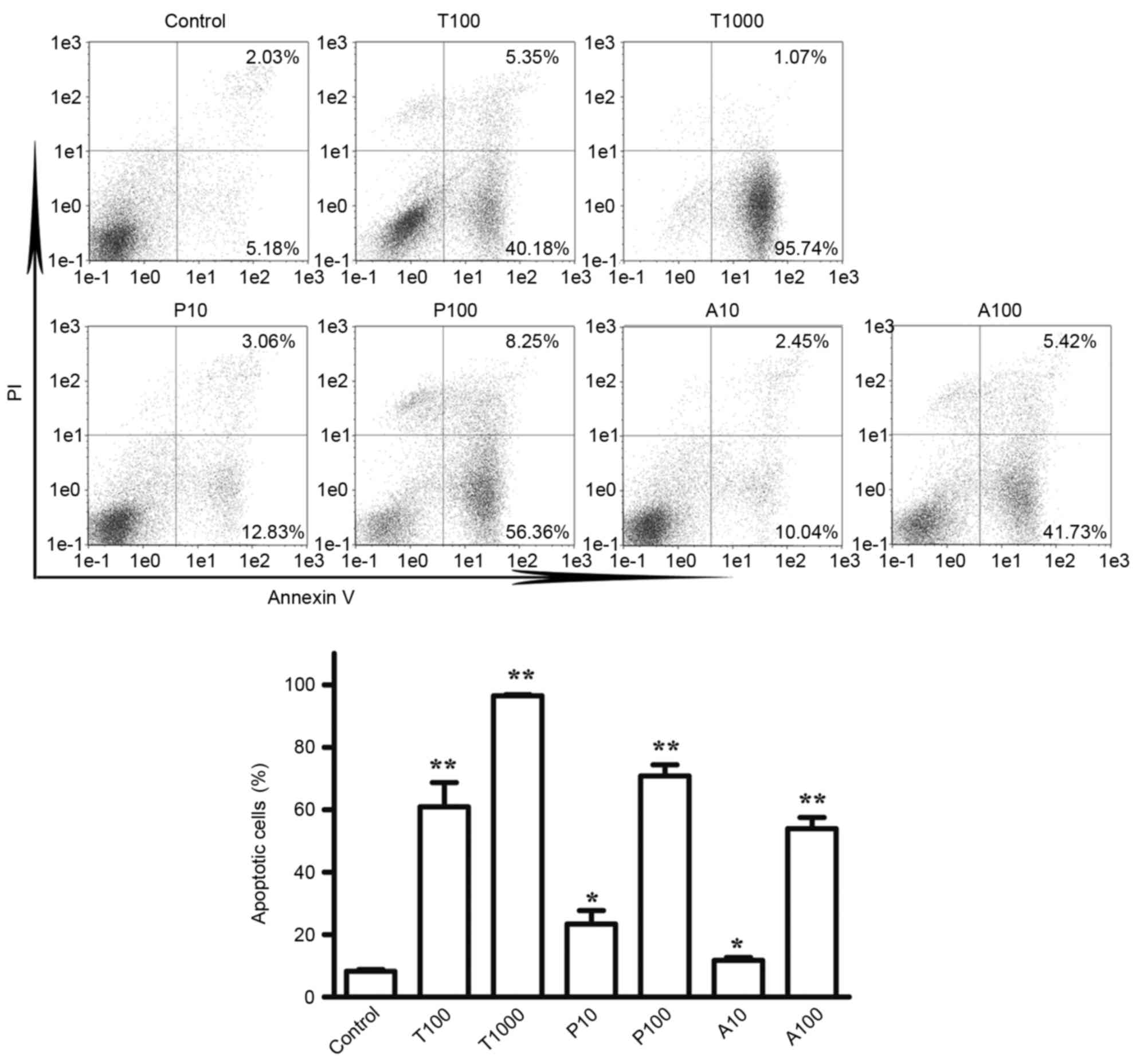
TKI258, ponatinib, and AZD4547 induce
the apoptosis of TPR-FGFR1-expressing Baf3 cells
To determine the effects of TKI258, ponatinib and
AZD4547 on the apoptosis of cells, Annexin V/PI staining was used.
According to the effects of the TKIs on cell proliferation, the
cells were treated with TKI258 at concentrations of 100 and 1,000
nmol/l, and with ponatinib and AZD4547 at concentrations of 10 and
100 nmol/l. As shown in Fig. 5A and
B, exposure to TKI258, ponatinib and AZD4547 at a concentration
of 100 nmol/l increased the apoptotic rate (Annexin V+/PI- and
Annexin+/PI+) from 8.28% in the control group to 60.90, 70.80 and
53.95% in the TPR-FGFR1-expressing Baf3 cells, respectively. No
significant difference was found among the three TKIs on their
effects on cell apoptosis (P>0.05). Exposure to TKI258 at a
concentration of 1,000 nmol/l markedly increased the apoptotic rate
to 96.81%.
TKI258, ponatinib and AZD4547 inhibit
the phosphorylation of TPR-FGFR1 and downstream signaling
molecules
The phosphorylation of FGFR1 leads to the activation
of four key downstream signaling pathways:
RAS-RAF-mitogen-activated protein kinase, phosphoinositide
3-kinase-Akt, STAT and PLCγ (12).
In the present study, western blot analysis was used to assess the
effects of TKI258, ponatinib and AZD4547 on the phosphorylation of
FGFR1, and the downstream signaling molecules, ERK1/2, Akt, STAT5
and PLCγ. The data (Fig. 6) showed
that 100 nmol/l ponatinib treatment resulted in a marked reduction
in the phosphorylation of ERK1/2, STAT5 and PLCγ, whereas the same
response required 1,000 nmol/l of TKI258. The effects of AZD4547 on
the activation of ERK1/2 and PLCγ were similar to the effects of
ponatimib, however, it had a more marked effect on the activation
of STAT5. Treatment with 10 nmol/l AZD4547 almost completely
inhibited the phosphorylation, whereas a 10-fold higher
concentration of ponatinib was required. The phosphorylation of Akt
was not affected by any of the TKIs.
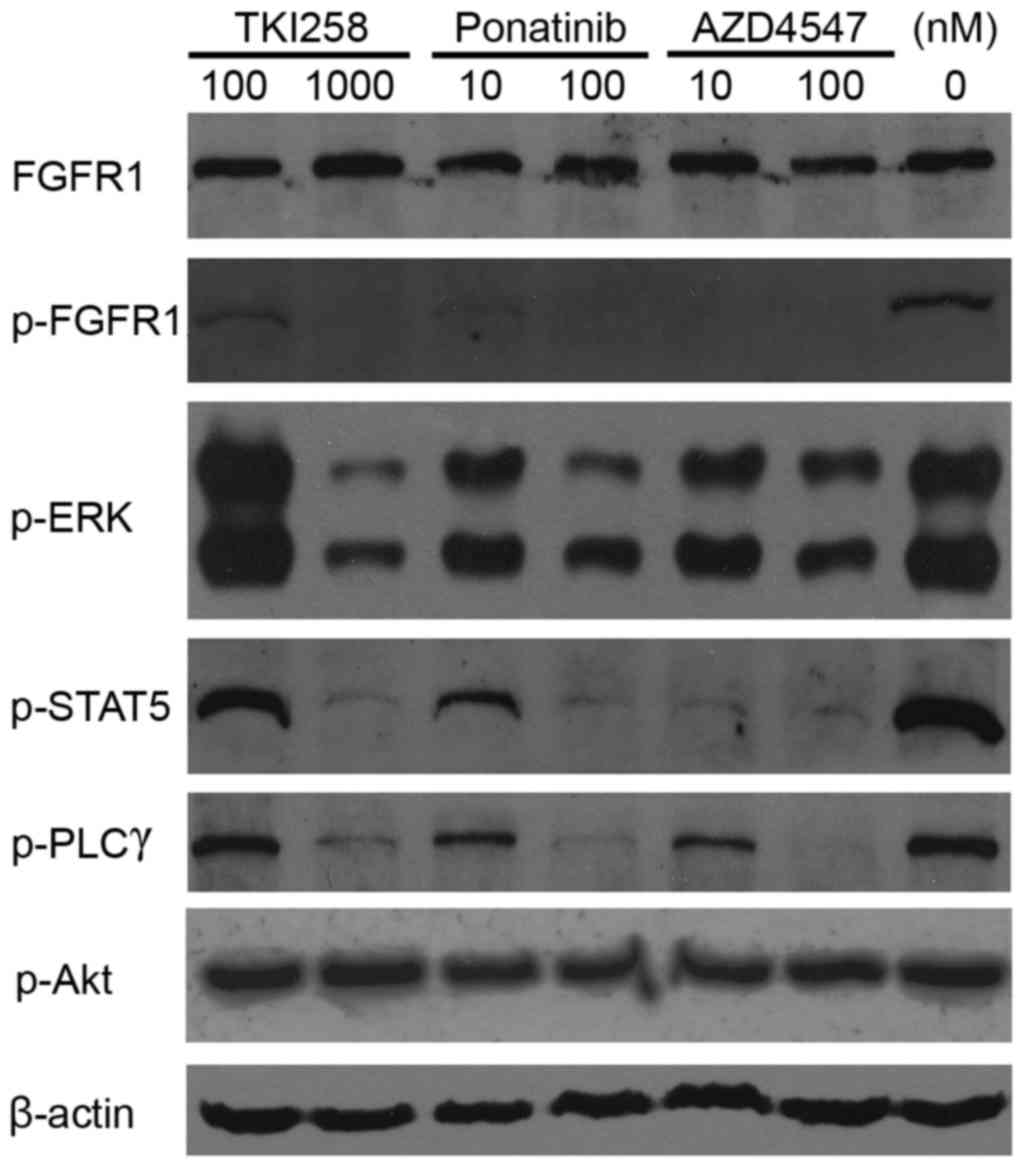 | Figure 6.Tytosine kinase inhibitors inhibit
FGFR1 activation. The TPR-FGFR1-expressing Baf3 cells were starved
overnight and treated with different concentrations of TKI258,
ponatinib or AZD4547 for 90 min. The phosphorylation of FGFR1, and
its downstream molecules, ERK, STAT5, PLCγ and Akt were detected
using western blot analysis. β-actin was used as a loading control.
TPR, translocated promoter region; FGFR1, fibroblast growth factor
receptor 1; ERK, extracellular signal-regulated kinase; STAT5,
signal transducer and activator of transcription; PLCγ,
phospholipase γ; p-, phosphorylated. |
Discussion
The TPR-FGFR1 fusion transcript was first
identified in our previous study in 2012 (2). To date, a total of four cases with
the t(1;8)(q25;p11.2) translocation have been reported (2,5,13,14).
All the patients showed myeloproliferative neoplasm bone marrow
morphology and peripheral monocytosis. Mammalian TPR is a 267-kDa
protein, which attaches to the nuclear pore complex (NPC) (15). It contains a large coiled-coil
forming amino-terminus, which involves the TprMet and NPC
association domain, and an acidic carboxyl-terminus with total
protein kinase and nuclear targeting activity (16,17).
In the present study, the TPR-FGFR1 fusion gene was stably
transfected into hematopoietic Baf3 cells. The resulting data
showed that the fusion gene transformed Baf3 cells into
IL3-independent cells, which was similar to other EMS fusion genes.
The present study also investigated the subcellular localization of
TPR-FGFR1, as Bangs et al (17) suggested that the subcellular
localization may affect the activity of the fusion protein. The
data from the present study showed that TPR-FGFR1 was localized
exclusively in the cytoplasm, rather than attaching to the NPC.
This was consistent with a previous study, in which the
NPC-associated domain and the nuclear targeting motif were
necessary for NPC attachment (16), whereas the latter motif was not
involved in TPR-FGFR.
Small molecule inhibitors targeting tyrosine kinase
activity have been developed for the treatment of various
malignancies. Particular success follows the use of imatinib for
the treatment of Philadelphia chromosome-positive chronic myeloid
leukemia (Ph+ CML) (18). As EMS is caused by the constitutive
activation of FGFR1, TKIs targeting FGFR1 are promising drugs for
the disease. TKI258, ponatinib and AZD4547 are three clinically
investigated FGFR inhibitors. Their effects have been evaluated in
patients with renal cell carcinoma, breast cancer, relapsed
multiple myeloma, urothelial cancer and chronic myelogenous
leukemia (19). However, none of
these TKIs have been investigated in patients with EMS. As TKI258,
ponatinib and AZD4547 are also efficient inhibitors against FGFR1,
they may be potential drugs for EMS. This has been partially
demonstrated in previous studies, which showed that TKI258,
ponatinib and AZD4547 inhibit the growth of ZNF198-FGFR1-,
BCR-FGFR1- and CUX-FGFR1-transformed Baf3 cells or
FGFR1OP2-FGFR1-positive KG1a cells (8–11).
However, the effects of these three TKIs have not been evaluated in
the same fusion gene systematically. In the present study, the
activities of TKI258, ponatinib and AZD4547 for EMS treatment were
compared using TPR-FGFR1-expressing Baf3 cells. The data showed
that, although all three TKIs markedly inhibited the proliferation
of TPR-FGFR1-expressing Baf3 cells, AZD4547 was the most efficient
drug, whereas TKI258 was the least efficient. The IC50
value of TKI258 was 112.374 nmol/l, which was ~10-fold higher,
compared with those of ponatinib and AZD4547. In addition, at the
concentration of 10 nmol/l, the rate of inhibition following
AZD4547 treatment was significantly higher, compared with that of
the other TKIs.
In order to confirm the effects of the three TKIs on
TPR-FGFR1, western blot analysis was used to determine their
activity against FGFR1 signaling. Similar results were obtained.
TKI258 was the least efficient drug for the activation of FGFR1 and
its downstream signaling molecules, ERK1/2, PLCg, and STAT5, as at
least 10-fold higher concentration than ponatinib and AZD4547 was
required to achieve the similar effect. Although the effects of
AZD4547 and ponatinib were similar on the activation of ERK1/2 and
PLCγ, AZD4547 was more potent in activating the phosphorylation of
STAT5. The treatment of cells with 10 nmol/l AZD4547 but minimal
ponatinib resulted in almost total inhibition of the activation of
STAT5. This was consistent with the proliferation data, which
showed that the inhibitory rate of 10 nmol/l AZD4547 was
significantly higher, compared with that of ponatinib at the same
concentration (66.5 vs. 39.7%, respectively; P<0.01). Taken
together, the results suggested that AZD4547 was the most efficient
drug for FGFR1 signaling, and the activation of STAT5 was a
critical event for TPR-FGFR1-mediated cell transformation.
Ponatinib is a non-selective TKI, which targets
breakpoint cluster region (BCR)-Abelson (ABL), FGFR, vascular
endothelial growth factor receptor (VEGFR) and platelet-derived
growth factor receptor, with similar enzyme IC50 values.
It is now approved for the treatment of CML or patients with
Ph+ acute lymphocytic leukemia with T315I
mutations, or those for whom all other TKIs have failed. The
indication of the drug has been narrowed due to severe
cardiovascular events; however, the mechanisms underlying these
adverse events remain to be fully elucidated. They may be induced
by BCR-ABL-specific effects, as cardiovascular adverse events have
also been identified in patients treated with other
BCR-ABL-targeted TKIs (20).
However, further investigations are required to verify this
possibility, as the majority of trials of ponatinib focus on CML
therapy, whereas adverse events in other malignancies remain to be
fully elucidated. The activity of ponatinib against VEGFR may be
another explanation for the adverse events. Previous studies have
indicated that VEGFR inhibitors increase the risk of pancreatitis
and treatment-associated mortality rates (21,22).
As a result, compared with ponatinib and TKI258, AZD4547 may be
safer for clinical use, as the specificity of AZD4547 eliminates
the adverse events associated with VEGFR.
A previous in vivo study showed that, despite
ponatinib inhibiting the proliferation of KG1a cells with an
IC50 value of 20 nmol/l, treatment with 30 mg/kg
ponatinib, which resulted in a mean plasma level of 561 nmol/l
following 6 h of drug administration, only prolonged the survival
rates of 40% of mice with xenograft KG1 cells (23). This suggests that treatment with a
single TKI may not be sufficient for EMS treatment. The data
obtained in the present study indicated that the activation of
another FGFR downstream signaling molecule, Akt, was not affected
by the three TKIs. Akt signaling remains active in hematological
malignancies, and is further activated by ZNF198-FGFR1 and
FOP-FGFR1 transformation (11,24–26).
Clinical trials have indicated that the combined therapy of Akt
inhibitors with bortezomib or lenalidomide significantly improves
the survival rates and responses of patients with multiple myeloma
(27,28). Therefore, combining TKIs and Akt
inhibitors may be a novel strategy for the treatment of EMS.
In conclusion, the data obtained in the present
study indicated that AZD4547 showed increased potency, compared
with TKI258 and ponatinib, on the inhibition of cell proliferation
and FGFR1 signaling. Considering the adverse events associated with
VEGFR inhibitors, AZD4547 may be the most promising drug for the
treatment of EMS.
References
|
1
|
Jackson CC, Medeiros LJ and Miranda RN:
8p11 myeloproliferative syndrome: A review. Hum Pathol. 41:461–476.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li F, Zhai YP, Tang YM, Wang LP and Wan
PJ: Identification of a novel partner gene, TPR, fused to FGFR1 in
8p11 myeloproliferative syndrome. Genes Chromosomes Cancer.
51:890–897. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gervais C, Dano L, Perrusson N, Hélias C,
Jeandidier E, Galoisy AC, Ittel A, Herbrecht R, Bilger K and
Mauvieux L: A translocation t(2;8)(q12;p11) fuses FGFR1 to a novel
partner gene, RANBP2/NUP358, in a
myeloproliferative/myelodysplastic neoplasm. Leukemia.
27:1186–1188. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nakamura Y, Ito Y, Wakimoto N, Kakegawa E,
Uchida Y and Bessho M: A novel fusion of SQSTM1 and FGFR1 in a
patient with acute myelomonocytic leukemia with t(5;8)(q35;p11)
translocation. Blood Cancer J. 4:e2652014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim SY, Kim JE, Park S and Kim HK:
Molecular identification of a TPR-FGFR1 fusion transcript in an
adult with myeloproliferative neoplasm, T-lymphoblastic lymphoma,
and a t(1;8)(q25;p11.2). Cancer Genet. 207:258–262. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Grand EK, Chase AJ, Heath C, Rahemtulla A
and Cross NC: Targeting FGFR3 in multiple myeloma: Inhibition of
t(4;14)-positive cells by SU5402 and PD173074. Leukemia.
18:962–966. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen J, Deangelo DJ, Kutok JL, Williams
IR, Lee BH, Wadleigh M, Duclos N, Cohen S, Adelsperger J, Okabe R,
et al: PKC412 inhibits the zinc finger 198-fibroblast growth factor
receptor 1 fusion tyrosine kinase and is active in treatment of
stem cell myeloproliferative disorder. Proc Natl Acad Sci USA.
101:14479–14484. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chase A, Grand FH and Cross NC: Activity
of TKI258 against primary cells and cell lines with FGFR1 fusion
genes associated with the 8p11 myeloproliferative syndrome. Blood.
110:3729–3734. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wasag B, Lierman E, Meeus P, Cools J and
Vandenberghe P: The kinase inhibitor TKI258 is active against the
novel CUX1-FGFR1 fusion detected in a patient with T-lymphoblastic
leukemia/lymphoma and t(7;8)(q22;p11). Haematologica. 96:922–926.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chase A, Bryant C, Score J and Cross NC:
Ponatinib as targeted therapy for FGFR1 fusions associated with the
8p11 myeloproliferative syndrome. Haematologica. 98:103–106. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gavine PR, Mooney L, Kilgour E, Thomas AP,
Al-Kadhimi K, Beck S, Rooney C, Coleman T, Baker D, Mellor MJ, et
al: AZD4547: An orally bioavailable, potent, and selective
inhibitor of the fibroblast growth factor receptor tyrosine kinase
family. Cancer Res. 72:2045–2056. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Turner N and Grose R: Fibroblast growth
factor signalling: From development to cancer. Nat Rev Cancer.
10:116–129. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yoshida C, Takeuchi M and Sadahira Y: A
novel t(1;8)(q25;p11.2) translocation associated with 8p11
myeloproliferative syndrome. Br J Haematol. 156:271–273. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim WS, Park SG, Park G, Jang SJ, Moon DS
and Kang SH: 8p11 myeloproliferative syndrome with
t(1;8)(q25;p11.2): A case report and review of the literature. Acta
Haematol. 133:101–105. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hase ME, Kuznetsov NV and Cordes VC: Amino
acid substitutions of coiled-coil protein Tpr abrogate anchorage to
the nuclear pore complex but not parallel, in-register
homodimerization. Mol Biol Cell. 12:2433–2452. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cordes VC, Hase ME and Muller L: Molecular
segments of protein Tpr that confer nuclear targeting and
association with the nuclear pore complex. Exp Cell Res. 245:43–56.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bangs P, Burke B, Powers C, Craig R,
Purohit A and Doxsey S: Functional analysis of Tpr: Identification
of nuclear pore complex association and nuclear localization
domains and a role in mRNA export. J Cell Biol. 143:1801–1812.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Giles FJ, Mauro MJ, Hong F, Ortmann CE,
McNeill C, Woodman RC, Hochhaus A, le Coutre PD and Saglio G: Rates
of peripheral arterial occlusive disease in patients with chronic
myeloid leukemia in the chronic phase treated with imatinib,
nilotinib, or non-tyrosine kinase therapy: A retrospective cohort
analysis. Leukemia. 27:1310–1315. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Katoh M and Nakagama H: FGF receptors:
Cancer biology and therapeutics. Med Res Rev. 34:280–300. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Loren CP, Aslan JE, Rigg RA, Nowak MS,
Healy LD, Gruber A, Druker BJ and McCarty OJ: The BCR-ABL inhibitor
ponatinib inhibits platelet immunoreceptor tyrosine-based
activation motif (ITAM) signaling, platelet activation and
aggregate formation under shear. Thromb Res. 135:155–160. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ghatalia P, Morgan CJ, Choueiri TK, Rocha
P, Naik G and Sonpavde G: Pancreatitis with vascular endothelial
growth factor receptor tyrosine kinase inhibitors. Crit Rev Oncol
Hematol. 94:136–145. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schutz FA, Je Y, Richards CJ and Choueiri
TK: Meta-analysis of randomized controlled trials for the incidence
and risk of treatment-related mortality in patients with cancer
treated with vascular endothelial growth factor tyrosine kinase
inhibitors. J Clin Oncol. 30:871–877. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ren M, Qin H, Ren R and Cowell JK:
Ponatinib suppresses the development of myeloid and lymphoid
malignancies associated with FGFR1 abnormalities. Leukemia.
27:32–40. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dong S, Kang S, Gu TL, Kardar S, Fu H,
Lonial S, Khoury HJ, Khuri F and Chen J: 14-3-3 integrates
prosurvival signals mediated by the AKT and MAPK pathways in
ZNF198-FGFR1-transformed hematopoietic cells. Blood. 110:360–369.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Guasch G, Ollendorff V, Borg JP, Birnbaum
D and Pébusque MJ: 8p12 stem cell myeloproliferative disorder: The
FOP-fibroblast growth factor receptor 1 fusion protein of the
t(6;8) translocation induces cell survival mediated by
mitogen-activated protein kinase and phosphatidylinositol
3-kinase/Akt/mTOR pathways. Mol Cell Biol. 21:8129–8142. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hideshima T, Catley L, Yasui H, Ishitsuka
K, Raje N, Mitsiades C, Podar K, Munshi NC, Chauhan D, Richardson
PG and Anderson KC: Perifosine, an oral bioactive novel
alkylphospholipid, inhibits Akt and induces in vitro and in vivo
cytotoxicity in human multiple myeloma cells. Blood. 107:4053–4062.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Richardson PG, Wolf J, Jakubowiak A,
Zonder J, Lonial S, Irwin D, Densmore J, Krishnan A, Raje N, Bar M,
et al: Perifosine plus bortezomib and dexamethasone in patients
with relapsed/refractory multiple myeloma previously treated with
bortezomib: Results of a multicenter phase I/II trial. J Clin
Oncol. 29:4243–4249. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jakubowiak AJ, Richardson PG, Zimmerman T,
Alsina M, Kaufman JL, Kandarpa M, Kraftson S, Ross CW, Harvey C,
Hideshima T, et al: Perifosine plus lenalidomide and dexamethasone
in relapsed and relapsed/refractory multiple myeloma: A phase I
multiple myeloma research consortium study. Br J Haematol.
158:472–480. 2012. View Article : Google Scholar : PubMed/NCBI
|