Introduction
Triple-negative breast cancer (TNBC) is an
aggressive subtype of breast cancer with a poor prognosis and high
mortality, which is diagnosed more frequently in young and
premenopausal women (1,2). Due to the absence of estrogen
receptors, progesterone receptor and the human epidermal growth
factor receptor-2 in TNBC tumor types, patients with TNBC do not
respond to hormone- or trastuzumab-based therapies, leaving
cytotoxic chemotherapy as the current therapy (3). Therefore, the identification of
critical genes involved in TNBC carcinogenesis may provide a
strategy for molecular target therapy of TNBC (4).
Microarray technology, which may be used to detect
the global gene expression, has provided an alternative method for
the molecular classification of different types of cancer, as well
as the exploration of potential prognostic biomarkers and
therapeutic targets. Based on microarray analysis, breast cancer
has been divided into distinctive subtypes according to different
gene expression patterns (5–11).
Furthermore, Rakha et al (4) performed a microarray analysis on a
relatively large set of 1,944 cases of invasive breast cancer that
contained TNBC tissues, as well as information on tumor size, lymph
node involvement and androgen receptor expression levels, and
determined these three parameters as the most useful
prognosticators of TNBC. Therefore, a systematic analysis of gene
expression patterns in TNBC may aid researchers to comprehend the
development and the treatment of this disease and to identify novel
therapeutic targets.
In the present study, biological microarray analysis
was used to analyze the gene expression profile of TNBC and to
screen the differentially expressed genes (DEGs) between TNBC and
adjacent normal tissues. Furthermore, altered biological pathways
in TNBC were identified using bioinformatics tools. Additionally, a
protein-protein interaction (PPI) network of DEGs was constructed
in order to identify the crucial genes involved in the process of
TNBC. The present study aimed to improve the understanding of the
underlying pathological mechanism and facilitate the discovery of
potential novel therapeutic targets for TNBC.
Materials and methods
Gene expression data
The GSE41970 gene expression profiles of TNBC were
downloaded from Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) of the National
Center for Biotechnology Information (Bethesda, MD, USA) based on
the GPL16299 platform data (NanoString nCounter mRNA Human Cancer
Reference Kit; NanoString, Inc., Ticino, Bellinzona, Switzerland)
(12). In total, 200 specimens,
including 160 primary TNBC specimens and 40 normal samples, were
used in the present study.
Data processing and differential
analysis
Qlucore Omics Explorer (QOE) software (version 3;
Qlucore AB, Lund, Sweden) was used to analyze the data from the
microarray (13,14). Intensity values of each probe-set
were log2 transformed, and a gene-specific t-test was
subsequently performed between TNBC samples and matched normal
samples. P<0.01 and [log2(fold change)>2] were
regarded as the cut-off criteria to screen out DEGs. To generate an
overview of the gene expression profile, hierarchical clustering
was also performed by QOE.
Gene ontology (GO) and pathway
enrichment analysis
To determine the biological pathways altered in
TNBC, GO biological process terms and Kyoto Encyclopedia of Genes
and Genomes (KEGG) pathway enrichment analyses were performed on
the DEGs using Database for Annotation, Visualization and
Integration Discovery (version 6.7; DAVID; http://david.abcc.ncifcrf.gov) (15). P<0.01 and false discovery rate
(FDR) <0.01 were set as the cut-off value.
Construction and analysis of PPI
network
It is possible to utilize PPI research to study DEG
protein functions at the molecular level, and cellular regulatory
mechanisms can be interpreted by elucidation of genome-wide protein
interactions (16). The PPI
network was constructed for the DEGs using information provided by
the Search Tool for the Retrieval of Interacting Genes, version 10
(STRING; http://string-db.org) (17), and was subsequently visualized
using Cytoscape version 3.2.1 (http://cytoscape.org) (18). The interactions of protein pairs
with a combined score >0.5 were retained in the network and the
hub genes were screened according to the degree score (the number
of neighbors). The subnetworks were then analyzed by Clustering
with Overlapping Neighborhood Expansion (ClusterONE; http://www.paccanarolab.org/clusterone).
GO functional and KEGG pathway enrichment analyses of the most
significant subnetworks were performed with a threshold of
P<0.01 and FDR<0.01.
Results
Identification of DEGs in TNBC
Using QOE, 121 DEGs were identified between 160 TNBC
and 40 normal tissues, including 101 upregulated genes and 20
downregulated genes. The 10 most significantly up- or downregulated
genes are listed in Table I.
 | Table I.Top 10 most significantly up- or
downregulated differentially expressed genes in triple-negative
breast cancer compared with normal tissue. |
Table I.
Top 10 most significantly up- or
downregulated differentially expressed genes in triple-negative
breast cancer compared with normal tissue.
| Gene symbol | P-value | Fold-change |
|---|
| Upregulated
genes |
|
|
|
BIRC5 |
6.50×10−36 | 48.63 |
|
MYBL2 |
4.32×10−34 | 57.33 |
|
TOP2A |
3.49×10−31 | 41.91 |
|
CDC2 |
4.07×10−29 | 32.75 |
|
MMP9 |
2.09×10−23 | 21.35 |
|
CHEK1 |
8.68×10−23 | 13.37 |
|
SPP1 |
2.21×10−22 | 40.41 |
|
TYMS |
4.26×10−22 | 18.21 |
|
E2F1 |
4.38×10−21 | 11.18 |
|
PCNA |
1.27×10−20 |
6.87 |
| Downregulated
genes |
|
|
|
IGFBP6 |
2.19×10−14 |
0.12 |
|
ESR1 |
1.09×10−13 |
0.09 |
|
DLC1 |
1.27×10−12 |
0.14 |
|
EGR1 |
6.87×10−11 |
0.20 |
|
IGF1 |
1.72×10−10 |
0.27 |
|
TGFBR3 |
3.37×10−10 |
0.16 |
|
PPARG |
3.84×10−10 |
0.23 |
|
NGFR |
3.93×10−10 |
0.12 |
|
CD34 |
6.67×10−9 |
0.27 |
|
FOS |
5.83×10−8 |
0.30 |
Clustering of DEGs
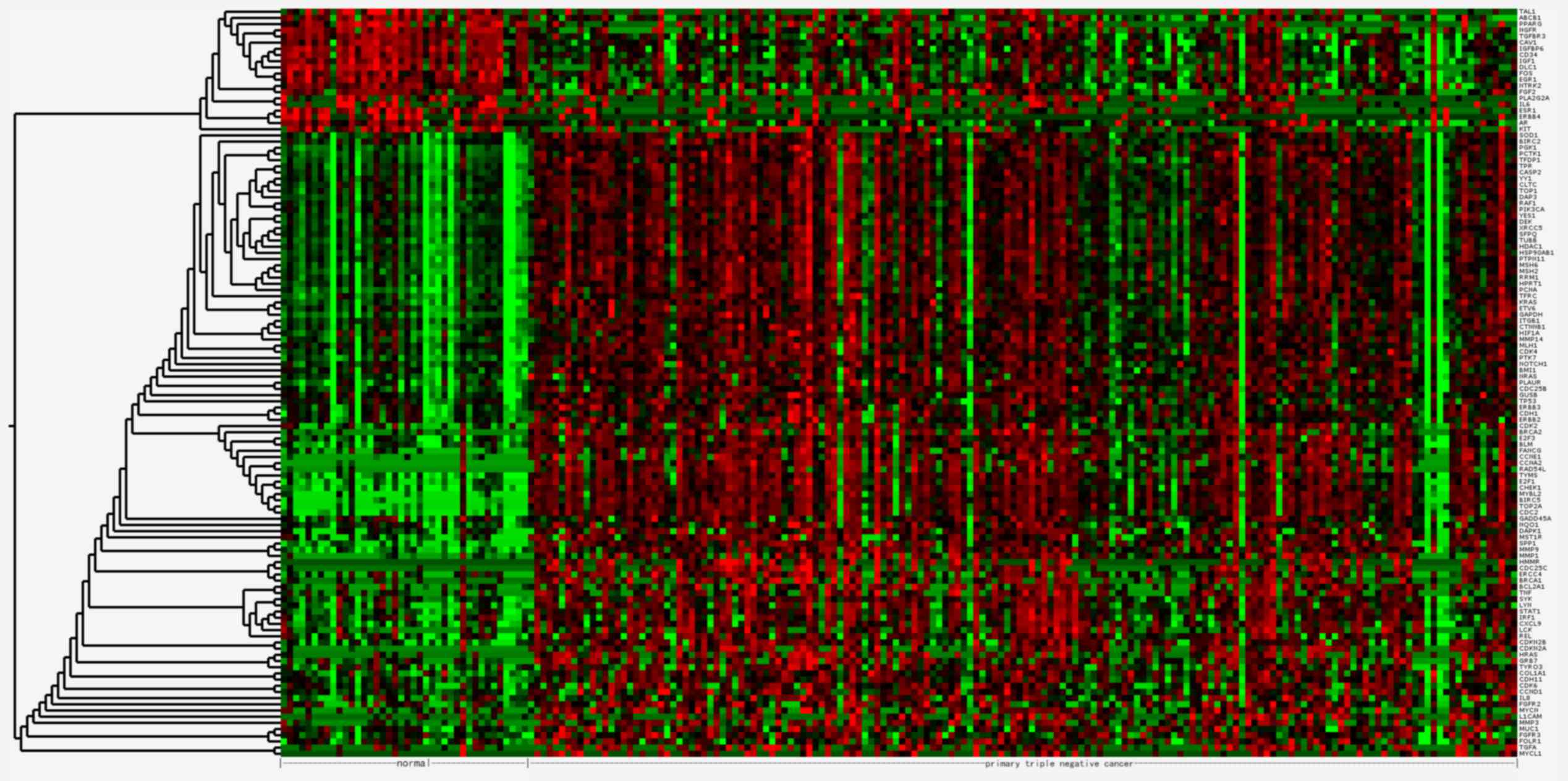
The 121 DEGs in TNBC compared with normal tissues
were selected for hierarchical clustering analysis. As presented in
Fig. 1, the DEGs were divided into
two major groups, separating 101 upregulated genes from 20
downregulated genes.
GO of biological process enrichment
analysis of up- and downregulated DEGs
The upregulated DEGs were significantly enriched in
83 biological processes and the top five significantly enriched
processes were mainly associated with the regulation of cell
proliferation, cell cycle and cell apoptosis (Table II). The downregulated DEGs were
significantly enriched in 18 biological processes, among which the
most significant were mainly involved in the regulation of cell
proliferation, macromolecule metabolic process and cell migration
(Table II).
| Table II.Top five significantly enriched
biology processes for differentially expressed genes. |
Table II.
Top five significantly enriched
biology processes for differentially expressed genes.
| GO ID | Term | Gene counts | P-value | FDR |
|---|
| Upregulated |
|
|
|
|
|
GO:0042127 | Regulation of cell
proliferation | 35 |
2.22×10−18 |
3.76×10−15 |
|
GO:0051726 | Regulation of cell
cycle | 24 |
8.20×10−17 |
1.89×10−13 |
|
GO:0008284 | Positive regulation
of cell proliferation | 26 |
8.35×10−17 |
1.89×10−13 |
|
GO:0042981 | Regulation of
apoptosis | 31 |
2.15×10−14 |
3.63×10−11 |
|
GO:0043067 | Regulation of
programmed cell death | 31 |
2.79×10−14 |
4.72×10−11 |
| Downregulated |
|
|
|
|
|
GO:0042127 | Regulation of cell
proliferation | 12 |
1.19×10−9 |
1.88×10−6 |
|
GO:0010604 | Positive regulation
of macromolecule metabolic process | 11 |
5.40×10−8 |
8.57×10−5 |
|
GO:0030334 | Regulation of cell
migration | 7 |
8.25×10−8 |
1.31×10−4 |
|
GO:0040012 | Regulation of
locomotion | 7 |
1.76×10−7 |
2.79×10−4 |
|
GO:0051270 | Regulation of cell
motion | 7 |
1.81×10−7 |
2.88×10−4 |
KEGG pathway enrichment analysis of
up- and downregulated DEGs
The upregulated DEGs were significantly enriched in
14 pathways, which mainly involved the cell cycle and pathways
associated with cancer, including the p53 signaling pathway
(Table III). In the
downregulated DEGs, four pathways were identified, however they
were without statistical significance (P>0.01 or
FDR>0.01).
| Table III.Significantly enriched pathways for
differentially expressed genes. |
Table III.
Significantly enriched pathways for
differentially expressed genes.
| KEGG ID | Term | Gene counts | P-value | FDR |
|---|
| hsa05200 | Pathways in
cancer | 37 |
3.77×10−24 |
4.19×10−21 |
| hsa05219 | Bladder cancer | 17 |
8.90×10−20 |
9.87×10−17 |
| hsa05223 | Non-small cell lung
cancer | 14 |
3.04×10−13 |
3.38×10−10 |
| hsa04110 | Cell cycle | 18 |
1.03×10−12 |
1.14×10−9 |
| hsa05215 | Prostate
cancer | 16 |
1.05×10−12 |
1.16×10−9 |
| hsa05212 | Pancreatic
cancer | 14 |
1.59×10−11 |
1.76×10−8 |
| hsa05220 | Chronic myeloid
leukemia | 14 |
2.74×10−11 |
3.04×10−8 |
| hsa05214 | Glioma | 13 |
5.32×10−11 |
5.91×10−8 |
| hsa05218 | Melanoma | 13 |
2.33×10−10 |
2.59×10−7 |
| hsa05213 | Endometrial
cancer | 11 |
2.22×10−9 |
2.46×10−6 |
| hsa05222 | Small cell lung
cancer | 12 |
2.30×10−8 |
2.56×10−5 |
| hsa05216 | Thyroid cancer | 8 |
1.25×10−7 |
1.39×10−4 |
| hsa05210 | Colorectal
cancer | 10 |
2.74×10−6 |
3.04×10−3 |
| hsa04115 | p53 signaling
pathway | 9 |
5.03×10−6 |
5.58×10−3 |
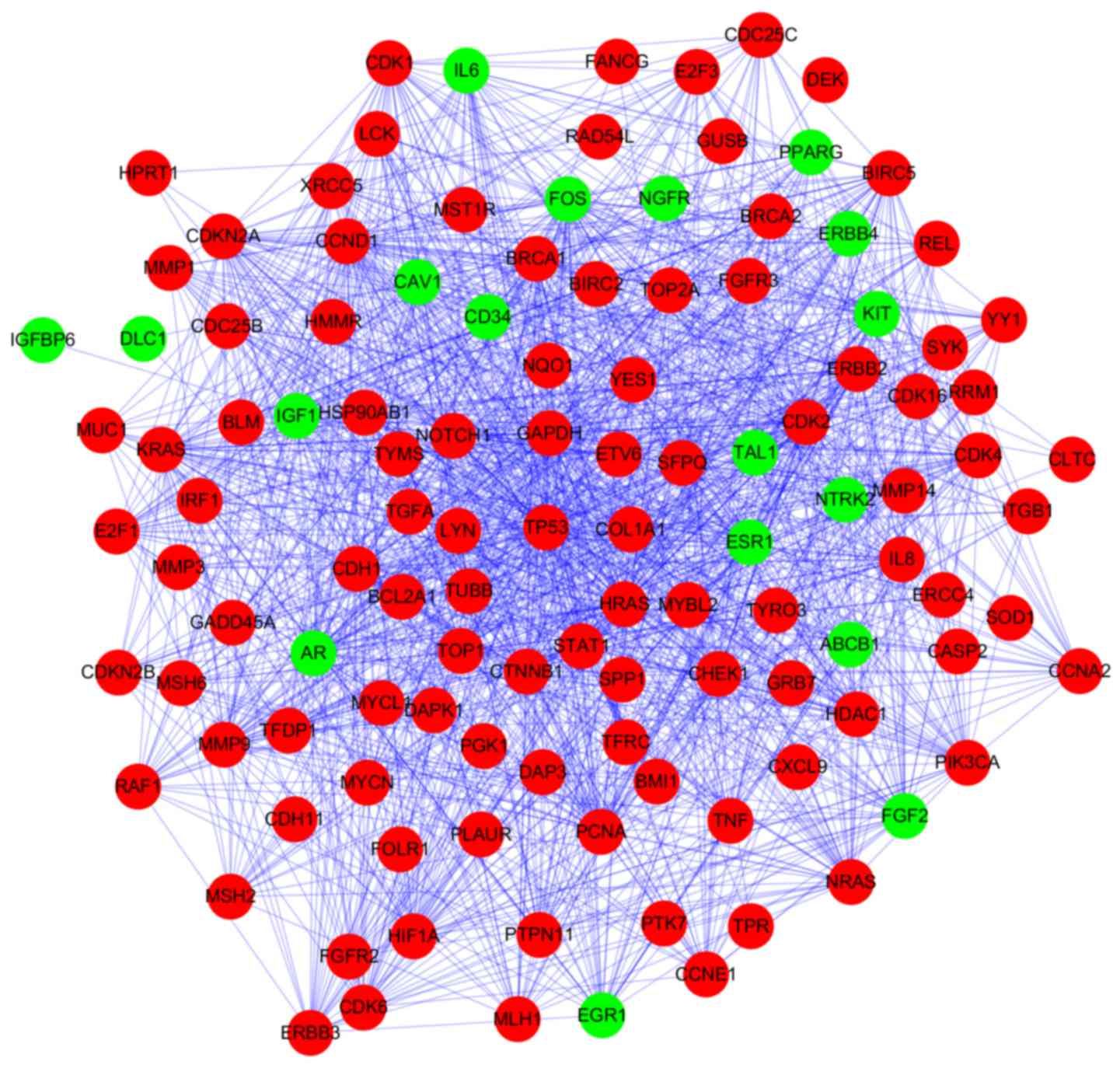
PPI network analysis
Based on STRING database analysis, a total of 1,264
protein pairs with the combined score >0.5 were identified. The
PPI network including 118 nodes and 1,264 edges was constructed
(Fig. 2) and the connectivity
degree of each node was calculated. The top five nodes TP53
(degrees, 86), glyceraldehyde-3-phosphate dehydrogenase (GAPDH;
degrees, 62), cyclin D1 (CCND1; degrees, 58), HRAS (degrees, 58)
and proliferating cell nuclear antigen (PCNA; degrees, 52) with
degrees >50 were screened as hub proteins in the PPI
network.
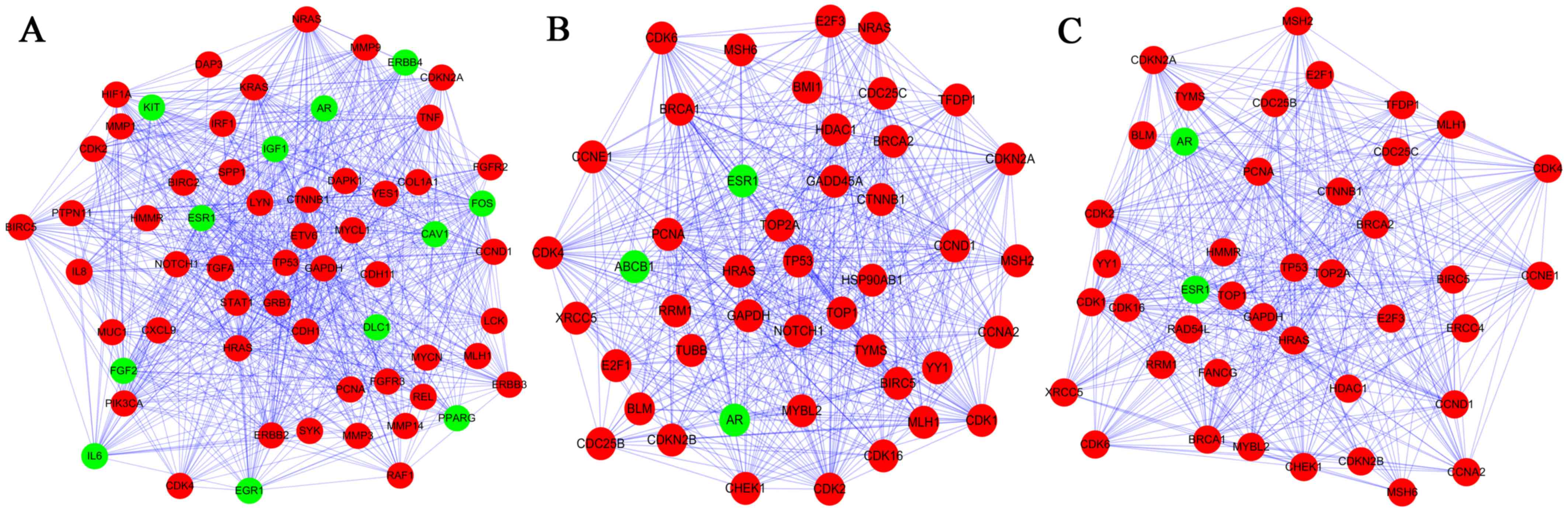
Three subnetworks (1,2 and 3) with P<0.05 were
established using the ClusterONE plugin (Fig. 3). The hub proteins TP53, GAPDH,
CCND1, HRAS and PCNA were demonstrated to be involved in all of
these three subnetworks. Subnetwork 1 was mainly associated with
regulation of cell proliferation and cell communication, while the
significant pathways were mainly associated with pathways in cancer
(Tables IV and V). By contrast, the other subnetworks
were mainly associated with the cell cycle (Table IV). Additionally, the significant
pathways correlated with subnetwork 2 and 3 were the cell cycle,
the p53 signaling pathway and pathways in cancer (Table V).
| Table IV.Top five significantly enriched
biology processes for differentially expressed genes in three
subnetworks. |
Table IV.
Top five significantly enriched
biology processes for differentially expressed genes in three
subnetworks.
| GO ID | Term | Gene counts | P-value | FDR |
|---|
| Subnetwork 1 |
|
|
|
|
|
GO:0042127 | Regulation of cell
proliferation | 30 |
3.15×10−20 |
5.31×10−17 |
|
GO:0010647 | Positive regulation
of cell communication | 21 |
7.11×10−18 |
1.20×10−14 |
|
GO:0009967 | Positive regulation
of signal transduction | 20 |
1.89×10−17 |
3.19×10−14 |
|
GO:0008284 | Positive regulation
of cell proliferation | 22 |
3.59×10−17 |
6.06×10−14 |
|
GO:0009719 | Response to
endogenous stimulus | 21 |
4.01×10−16 |
7.44×10−13 |
| Subnetwork 2 |
|
|
|
|
|
GO:0022402 | Cell cycle
process | 24 |
1.05×10−20 |
1.65×10−17 |
|
GO:0007049 | Cell cycle | 26 |
3.03×10−20 |
4.79×10−17 |
|
GO:0051726 | Regulation of cell
cycle | 19 |
3.49×10−18 |
5.51×10−15 |
|
GO:0022403 | Cell cycle
phase | 20 |
7.57×10−18 |
1.20×10−14 |
|
GO:0051329 | Interphase of
mitotic cell cycle | 13 |
3.33×10−16 |
5.22×10−13 |
| Subnetwork 3 |
|
|
|
|
|
GO:0022402 | Cell cycle
process | 23 |
1.66×10−20 |
2.59×10−17 |
|
GO:0007049 | Cell cycle | 25 |
2.88×10−20 |
4.51×10−17 |
|
GO:0051726 | Regulation of cell
cycle | 19 |
4.22×10−19 |
6.61×10−16 |
|
GO:0022403 | Cell cycle
phase | 19 |
2.27×10−17 |
3.55×10−14 |
|
GO:0006259 | DNA metabolic
process | 20 |
3.35×10−17 |
5.25×10−14 |
| Table V.Top five significantly enriched
pathways for differentially expressed genes in three
subnetworks. |
Table V.
Top five significantly enriched
pathways for differentially expressed genes in three
subnetworks.
| KEGG ID | Term | Gene counts | P-value | FDR |
|---|
| Subnetwork 1 |
|
|
|
|
|
hsa05200 | Pathways in
cancer | 32 |
3.04×10−25 |
3.30×10−22 |
|
hsa05219 | Bladder cancer | 15 |
4.66×10−19 |
5.05×10−16 |
|
hsa05215 | Prostate
cancer | 14 |
1.23×10−12 |
1.34×10−9 |
|
hsa05213 | Endometrial
cancer | 11 |
3.80×10−11 |
4.12×10−8 |
|
hsa05218 | Melanoma | 12 |
4.29×10−11 |
4.65×10−8 |
| Subnetwork 2 |
|
|
|
|
|
hsa04110 | Cell cycle | 19 |
3.60×10−20 |
3.72×10−17 |
|
hsa05200 | Pathways in
cancer | 21 |
5.11×10−15 |
5.27×10−12 |
|
hsa05215 | Prostate
cancer | 11 |
3.82×10−10 |
3.94×10−7 |
|
hsa04115 | p53 signaling
pathway | 10 |
7.34×10−10 |
7.57×10−7 |
|
hsa05220 | Chronic myeloid
leukemia | 10 |
1.81×10−9 |
1.86×10−6 |
| Subnetwork 3 |
|
|
|
|
|
hsa04110 | Cell cycle | 18 |
2.32×10−19 |
2.37×10−16 |
|
hsa05200 | Pathways in
cancer | 19 |
2.03×10−13 |
2.07×10−10 |
|
hsa04115 | p53 signaling
pathway | 9 |
9.25×10−9 |
9.43×10−6 |
|
hsa05220 | Chronic myeloid
leukemia | 9 |
2.04×10−8 |
2.08×10−5 |
|
hsa05223 | Non-small cell lung
cancer | 8 |
4.40×10−8 |
4.49×10−5 |
Discussion
TNBC is one of the most deadly breast cancer
subtypes due to the lack of an effective treatment. The improvement
of the diagnosis and therapeutic methods of this disease relies on
the discovery of novel potential molecular markers. In the present
study, a total of 121 DEGs between TNBC and normal tissues were
identified, consisting of 101 upregulated and 20 downregulated
genes. With 121 gene signatures mapped from the STRING database, a
giant component PPI network was established with 118 nodes and
1,264 edges. After applying the ClusterONE clustering algorithm,
three significant subnetworks with highly connected nodes were
obtained. The top five ranked genes as hub nodes with maximum
degrees were identified, including TP53, GAPDH, CCND1, HRAS and
PCNA. In addition, it was revealed that these hub nodes existed in
all of the three subnetworks, which contributed to the biological
processes of cell growth and the cell cycle and connected the cell
cycle, p53 signaling pathway and pathways in cancer.
TP53, also termed p53, encodes a tumor suppressor
protein that controls the cell cycle arrest, apoptosis, senescence,
DNA repair and changes in metabolism (19). Mutations in this gene are
associated with a variety of human cancer types, including TNBC,
lung cancer and high-grade serous ovarian tumor types (20). p53 is mutated and overexpressed in
~25–30% of human breast cancer (21), and had an increased incidence in
TNBC (22,23). Nishimura et al (24) also found that high proliferation
rate and frequent p53 overexpression occurred in TNBC. Furthermore,
p53 is also expressed as smaller isoforms, some of which inhibit
wild-type p53. Avery-Kiejda et al (25) reported that Δ40p53, one of the p53
isoforms, was significantly upregulated in tumor tissue when
compared with the normal breast and was closely associated with
TNBC. Mutation and isoforms of p53 may provide an alternate
explanation for the malfunction of p53 pathway and deregulated p53
signaling in TNBC. The present study demonstrated that the p53 gene
was elevated in the TNBC samples and that p53 was a hub protein
with a degree score of 86 in the established PPI network.
Therefore, the p53 gene may be a key regulator in TNBC
development.
GAPDH, originally identified as a glycolytic enzyme
is considered a housekeeping gene. It is frequently used as an
internal standard for gene expression in RNA or protein
experiments. However, the abnormal expression of GAPDH has been
confirmed to have a close association with various types of cancer
(26), as it serves an important
role in carcinogenesis and cell death, as well as energy metabolism
(27). Notably, increased levels
of GAPDH were observed in most types of human cancer and were often
correlated with reduced survival (28,29).
Results of the present study indicated that GAPDH is predicated as
a pivotal gene associated with TNBC and the cell cycle, and may be
a potential biomarker for detection and prevention of TNBC.
CCND1, a member of the cyclin-dependent kinase
regulator family, is required for the activation of CDK4 and CDK6,
and is recognized as a critical modulator of the cell cycle and a
positive regulator of cell proliferation (30). Aberrant amplification and
overexpression of CCND1 are a driving force in 13–20% of human
breast cancer, and are associated with poor disease outcome
(31). However, Mylona et
al (32) suggested that CCND1
overexpression may serve as a marker for prolonged survival of
patients with TNBC. Although the exact role of CCND1 in TNBC
remains controversial, according to the findings of the present
study, CCND1 overexpression was detected in TNBC tissues, and it
acted as a hub gene in the PPI network of TNBC. These data implied
that CCND1 is of clinical importance for TNBC.
HRAS, a member of the ras superfamily of genes,
encodes for a 21-kDa protein (p21), which takes part in the signal
transduction pathways that control proliferation and apoptosis, and
regulate the cell cycle. Ras genes are involved in a wide variety
of human tumor types and there is a known positive correlation
between HRAS activation and breast cancer (33). In addition, previous studies
(34,35) have shown that HRAS can induce
invasion and migration of the breast cancer cell line, MCF10A. From
the present study, HRAS may serve a significant role in the
pathogenesis of TNBC and, given its functional significance in
various types of cancer, it may also be a potential therapeutic
target in the treatment of TNBC.
PCNA is well known as a coordinator of essential
cellular processes for cell growth, death, and maintenance.
Accumulated evidence suggests that an enhanced level of PCNA often
correlates with carcinogenesis and that PCNA levels can be used as
a prognostic marker in certain cases (36–39).
Yu et al (40) demonstrated
that when PCNA activity was inhibited by a peptide, it suppressed
the growth of TNBC cells. Given the critical function of PCNA in
cancer growth, it is possible that the targeting of PCNA may be a
viable therapeutic method for TNBC.
Pathways in cancer and pathways involving the cell
cycle were demonstrated to be highly enriched in the DEGs for the
PPI network and the subnetworks. All these pathways have been
implicated in the development of breast cancer. Notably, the most
significantly enriched pathways were pathways in cancer, which
refers to some classical cancer pathways, including p53 signaling,
Wnt signaling, and Janus kinase-signal transducer and activator of
transcription (JAK-STAT) signaling pathways. Certain previous
studies (41–43) suggested that these critical
signaling nodes, including p53, β-catenin and JAK/STAT3, can be
used as therapeutic targets for the treatment of TNBC. In addition,
cancer has been viewed as a cell cycle disease (44). Substantial evidence has indicated
that the hub genes, P53, CCND1, HRAS and PCNA, are related to the
pathways of the cell cycle, which is in accordance with the
functions of networks identified in the present study.
In conclusion, 121 genes were revealed to be
differentially expressed between TNBC and normal tissues by
integrated analysis. Among them were TP53, GAPDH, CCND1, HRAS and
PCNA, and these may be involved in TNBC progression via the cell
cycle pathways and pathways in cancer. These findings may improve
the understanding of the pathogenesis of TNBC and the development
of targeted treatments for TNBC. Further experiments are required
to confirm the findings of this work and the hypotheses put
forward.
Acknowledgements
The current research was supported by the Institute
of Genetic Engineering at the Southern Medical University
(Guangzhou, China) and the present study was funded by the National
Natural Science Foundation of China (grant no. 39880032), the
Guangdong Foundation for Leading Talented Scientists (grant no.
C1030925) and the China Postdoctoral Science Foundation Funded
Project (grant no. 2016M602487). The authors would like to
acknowledge the excellent technical assistance of Dr Kaiyuan Ji
(School of Basic Medicine, Southern Medical University, China) and
Dr Zijun Qiao (Institutes of Biomedical Sciences, Fudan University,
China) during the present study.
References
|
1
|
Dawson SJ, Provenzano E and Caldas C:
Triple negative breast cancers: Clinical and prognostic
implications. Eur J Cancer. 45:(Suppl 1). S27–S40. 2009. View Article : Google Scholar
|
|
2
|
Reis-Filho JS and Tutt AN: Triple negative
tumours: A critical review. Histopathology. 52:108–118. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Anders CK and Carey LA: Biology,
metastatic patterns and treatment of patients with triple-negative
breast cancer. Clin Breast Cancer. 9:(Suppl 2). S73–S81. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rakha EA, El-Sayed ME, Green AR, Lee AH,
Robertson JF and Ellis IO: Prognostic markers in triple-negative
breast cancer. Cancer. 109:25–32. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kapp AV, Jeffrey SS, Langerød A,
Børresen-Dale AL, Han W, Noh DY, Bukholm IR, Nicolau M, Brown PO
and Tibshirani R: Discovery and validation of breast cancer
subtypes. BMC Genomics. 7:2312006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mattie MD, Benz CC, Bowers J, Sensinger K,
Wong L, Scott GK, Fedele V, Ginzinger D, Getts R and Haqq C:
Optimized high-throughput microRNA expression profiling provides
novel biomarker assessment of clinical prostate and breast cancer
biopsies. Mol Cancer. 5:242006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hu Z, Fan C, Oh DS, Marron JS, He X,
Qaqish BF, Livasy C, Carey LA, Reynolds E, Dressler L, et al: The
molecular portraits of breast tumors are conserved across
microarray platforms. BMC Genomics. 7:962006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
El-Rehim Abd DM, Ball G, Pinder SE, Rakha
E, Paish C, Robertson JF, Macmillan D, Blamey RW and Ellis IO:
High-throughput protein expression analysis using tissue microarray
technology of a large well-characterised series identifies
biologically distinct classes of breast cancer confirming recent
cDNA expression analyses. Int J Cancer. 116:340–350. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sorlie T, Tibshirani R, Parker J, Hastie
T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, et
al: Repeated observation of breast tumor subtypes in independent
gene expression data sets. Proc Natl Acad Sci USA. 100:8418–8423.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sorlie T, Perou CM, Tibshirani R, Aas T,
Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey
SS, et al: Gene expression patterns of breast carcinomas
distinguish tumor subclasses with clinical implications. Proc Natl
Acad Sci USA. 98:10869–10874. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Perou CM, Sørlie T, Eisen MB, van de Rijn
M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA,
et al: Molecular portraits of human breast tumours. Nature.
406:747–752. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cascione L, Gasparini P, Lovat F, Carasi
S, Pulvirenti A, Ferro A, Alder H, He G, Vecchione A, Croce CM, et
al: Integrated microRNA and mRNA signatures associated with
survival in triple negative breast cancer. PLoS One. 8:e559102013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ma H, Morey R, O'Neil RC, He Y, Daughtry
B, Schultz MD, Hariharan M, Nery JR, Castanon R, Sabatini K, et al:
Abnormalities in human pluripotent cells due to reprogramming
mechanisms. Nature. 511:177–183. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Killian JK, Kim SY, Miettinen M, Smith C,
Merino M, Tsokos M, Quezado M, Smith WI Jr, Jahromi MS, Xekouki P,
et al: Succinate dehydrogenase mutation underlies global epigenomic
divergence in gastrointestinal stromal tumor. Cancer Discov.
3:648–657. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sherman BT, da Huang W, Tan Q, Guo Y, Bour
S, Liu D, Stephens R, Baseler MW, Lane HC and Lempicki RA: DAVID
Knowledgebase: A gene-centered database integrating heterogeneous
gene annotation resources to facilitate high-throughput gene
functional analysis. BMC Bioinformatics. 8:4262007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li S, Armstrong CM, Bertin N, Ge H,
Milstein S, Boxem M, Vidalain PO, Han JD, Chesneau A, Hao T, et al:
A map of the interactome network of the metazoan C. elegans.
Science. 303:540–543. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43:D447–D452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Freed-Pastor WA and Prives C: Mutant p53:
One name, many proteins. Genes Dev. 26:1268–1286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kandoth C, McLellan MD, Vandin F, Ye K,
Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al:
Mutational landscape and significance across 12 major cancer types.
Nature. 502:333–339. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tennis M, Krishnan S, Bonner M, Ambrosone
CB, Vena JE, Moysich K, Swede H, McCann S, Hall P, Shields PG and
Freudenheim JL: p53 Mutation analysis in breast tumors by a DNA
microarray method. Cancer Epidemiol Biomarkers Prev. 15:80–85.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hanby AM: Aspects of molecular phenotype
and its correlations with breast cancer behaviour and taxonomy. Br
J Cancer. 92:613–617. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li XR, Liu M, Zhang YJ, Wang JD, Zheng YQ,
Li J, Ma B and Song X: CK5/6, EGFR, Ki-67, cyclin D1, and nm23-H1
protein expressions as predictors of pathological complete response
to neoadjuvant chemotherapy in triple-negative breast cancer
patients. Med Oncol. 28:(Suppl 1). S129–S134. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nishimura R and Arima N: Is triple
negative a prognostic factor in breast cancer? Breast Cancer.
15:303–308. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Avery-Kiejda KA, Morten B, Wong-Brown MW,
Mathe A and Scott RJ: The relative mRNA expression of p53 isoforms
in breast cancer is associated with clinical features and outcome.
Carcinogenesis. 35:586–596. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Caradec J, Sirab N, Revaud D, Keumeugni C
and Loric S: Is GAPDH a relevant housekeeping gene for
normalisation in colorectal cancer experiments? Br J Cancer.
103:1475–1476. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Colell A, Green DR and Ricci JE: Novel
roles for GAPDH in cell death and carcinogenesis. Cell Death
Differ. 16:1573–1581. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Altenberg B and Greulich KO: Genes of
glycolysis are ubiquitously overexpressed in 24 cancer classes.
Genomics. 84:1014–1020. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guo C, Liu S and Sun MZ: Novel insight
into the role of GAPDH playing in tumor. Clin Transl Oncol.
15:167–172. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hadzisejdić I, Mustać E, Jonjić N,
Petković M and Grahovac B: Nuclear EGFR in ductal invasive breast
cancer: Correlation with cyclin-D1 and prognosis. Mod Pathol.
23:392–403. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Courjal F, Cuny M, Simony-Lafontaine J,
Louason G, Speiser P, Zeillinger R, Rodriguez C and Theillet C:
Mapping of DNA amplifications at 15 chromosomal localizations in
1875 breast tumors: Definition of phenotypic groups. Cancer Res.
57:4360–4367. 1997.PubMed/NCBI
|
|
32
|
Mylona E, Tzelepis K, Theohari I,
Giannopoulou I, Papadimitriou C and Nakopoulou L: Cyclin D1 in
invasive breast carcinoma: Favourable prognostic significance in
unselected patients and within subgroups with an aggressive
phenotype. Histopathology. 62:472–480. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Watson DM, Elton RA, Jack WJ, Dixon JM,
Chetty U and Miller WR: The H-ras oncogene product p21 and
prognosis in human breast cancer. Breast Cancer Res Treat.
17:161–169. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Moon A, Kim MS, Kim TG, Kim SH, Kim HE,
Chen YQ and Kim HR: H-ras, but not N-ras, induces an invasive
phenotype in human breast epithelial cells: A role for MMP-2 in the
H-ras-induced invasive phenotype. Int J Cancer. 85:176–181. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yong HY, Hwang JS, Son H, Park HI, Oh ES,
Kim HH, Kim DK, Choi WS, Lee BJ, Kim HR and Moon A: Identification
of H-Ras-specific motif for the activation of invasive signaling
program in human breast epithelial cells. Neoplasia. 13:98–107.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Malkas LH, Herbert BS, Abdel-Aziz W,
Dobrolecki LE, Liu Y, Agarwal B, Hoelz D, Badve S, Schnaper L,
Arnold RJ, et al: A cancer-associated PCNA expressed in breast
cancer has implications as a potential biomarker. Proc Natl Acad
Sci USA. 103:19472–19477. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bukholm IR, Bukholm G, Holm R and Nesland
JM: Association between histology grade, expression of HsMCM2, and
cyclin A in human invasive breast carcinomas. J Clin Pathol.
56:368–373. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee JS, Kim HS, Jung JJ, Kim YB, Park CS
and Lee MC: Correlation between angiogenesis, apoptosis and cell
proliferation in invasive ductal carcinoma of the breast and their
relation to tumor behavior. Anal Quant Cytol Histol. 23:161–168.
2001.PubMed/NCBI
|
|
39
|
Aziz SA, Pervez S, Khan SM, Kayani N and
Nasir MI: Prognostic value of proliferating cell nuclear antigen
(PCNA) in infiltrating ductal carcinoma breast. J Coll Physicians
Surg Pak. 15:225–229. 2005.PubMed/NCBI
|
|
40
|
Yu YL, Chou RH, Liang JH, Chang WJ, Su KJ,
Tseng YJ, Huang WC, Wang SC and Hung MC: Targeting the EGFR/PCNA
signaling suppresses tumor growth of triple-negative breast cancer
cells with cell-penetrating PCNA peptides. PLoS One. 8:e613622013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Turner N, Moretti E, Siclari O, Migliaccio
I, Santarpia L, D'Incalci M, Piccolo S, Veronesi A, Zambelli A, Del
Sal G and Di Leo A: Targeting triple negative breast cancer: Is p53
the answer? Cancer Treat Rev. 39:541–550. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dey N, Barwick BG, Moreno CS,
Ordanic-Kodani M, Chen Z, Oprea-Ilies G, Tang W, Catzavelos C,
Kerstann KF, Sledge GW Jr, et al: Wnt signaling in triple negative
breast cancer is associated with metastasis. BMC Cancer.
13:5372013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Poage GM, Hartman ZC and Brown PH:
Revealing targeted therapeutic opportunities in triple-negative
breast cancers: A new strategy. Cell Cycle. 12:2705–2706. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sherr CJ: Cancer cell cycles. Science.
274:1672–1677. 1996. View Article : Google Scholar : PubMed/NCBI
|