Introduction
Tuberculosis (TB) is caused by Mycobacterium
tuberculosis infection and remains one of the major infectious
diseases affecting people worldwide, particularly in developing
countries. The morbidity and mortality of TB has been exacerbated
by the human immunodeficiency virus, the emergence of
multidrug-resistant TB, and extensively drug-resistant strains of
M. tuberculosis (1).
Therefore, it is important to understand the disease and to
investigate novel anti-TB drugs.
Unlike the synthesis of cysteine from methionine
through homocysteine in fungi and animals (2), serine acetyltransferase (CysE)
catalyzes the conversion of acetyl-coenzyme A (CoA) and L-serine to
CoA and O-acetyl-L-serine, which is the first product and a
key amino acid in the biosynthesis of L-cysteine in microorganisms
and plants (3,4). Cysteine is required for the
biosynthesis of essential compounds, including L-methionine,
thiamine, biotin, coenzyme A and other sulfur-containing compounds.
Notably, CysE has no known biological function in humans, as
different pathways are used in the anabolism of cysteine in
microorganisms and humans. The M. tuberculosis CysE is
therefore considered as a potential anti-TB drug target (5,6). Our
previous study demonstrated that the Rv2335 protein was the M.
tuberculosis homolog of CysE, encoded by the cysE gene,
and determined and described the kinetic parameters and optimal
catalytic conditions of the enzyme (7). However, to the best of our knowledge,
there is currently no information regarding the structure and
active sites of M. tuberculosis CysE. Determination of the
protein structure is crucial for investigating the relationship
between structure and function of any protein. Homology modeled
structures may characterize conserved domains and active amino
acids, which are essential to the function of the protein.
In the present study, the CysE amino acid sequence
from five species, M. tuberculosis, Escherichia coli, Salmonella
typhimurium, Haemophilus influenza and Entamoeba
histolytica, was compared. This comparison allowed for the
prediction of the secondary structure of M. tuberculosis
CysE and the construction of a 3D model. Three residues, D67, H82
and H117, were identified as significant sites for activity. Such
identifications and characterizations may facilitate screening for
enzyme inhibitors to catalytic processes in M. tuberculosis,
and thus therapy for patients with TB.
Materials and methods
Materials
Reagents and enzymes involved in gene cloning and
site-directed mutagenesis were purchased from Takara Bio, Inc.
(Otsu, Japan). The chemicals for all enzyme assays were purchased
from Sigma-Aldrich (Merck Millipore, Darmstadt, Germany). The
Ni-NTA agarose column used for protein purification was obtained
from Qiagen GmbH (Hilden, Germany).
Sequence analysis and alignments
The amino acid sequences of M. tuberculosis
CysE (NP_216851) were retrieved from the National Center for
Biotechnology Information (NCBI) GenBank database (Bethesda, MD,
USA) (8). The ProtParam tool in
the ExPASy server (http://web.expasy.org/protparam) was used to predict
the physiochemical properties of M. tuberculosis CysE
(9). The parameters included:
Molecular weight, theoretical isoelectric point (pI), amino acid
composition, atomic composition, extinction coefficient, estimated
half-life, instability index, aliphatic index and grand average of
hydropathy (GRAVY). CysE protein sequences of M.
tuberculosis CysE, E. coli CysE (AJF44936), S.
typhimurium CysE (KPF36926.1), H. influenzae CysE
(KMZ31837.1) and E. histolytica CysE (BAA82868.1) were
aligned using the MultAlin sequence alignment tool (http://multalin.toulouse.inra.fr/multalin) (10).
Secondary structure prediction and
functional annotation of the M. tuberculosis CysE protein
Based on sequence similarity searches with
orthologous family members, several bioinformatics tools were used
to analyze the conserved domains of CysE: The European
Bioinformatics Institutes' (EBI) InterProScan tool, which combines
different protein signature recognition methods from InterPro
consortium member databases into one resource (11); the University College London's
PSIPRED protein structure prediction server (http://bioinf.cs.ucl.ac.uk/psipred), which is based on
a neural network algorithm for predicting structural information
about a protein from its amino acid sequence alone (12); and the NCBI Conserved Domains
Database (NCBI-CDD; https://www.ncbi.nlm.nih.gov/cdd), which is a protein
annotation resource that consists of a collection of well-annotated
multiple sequence alignment models for ancient domains and
full-length proteins (13).
3D modeling of CysE protein
A 3D structure of the M. tuberculosis CysE
protein was modeled by comparative protein modeling methods using
the SWISS-MODEL server (http://swissmodel.expasy.org) (14). A suitable template for homology
modeling was selected using NCBI Position Specific Iterated-Basic
Local Alignment Search Tool (PSI-BLAST) (15) against the Research Collaboratory
for Structural Bioinformatics Protein Data Bank (PDB) (http://rcsb.org). The ExPASy Swiss-PdbViewer 3.5
(http://spdbv.vital-it.ch) was used to produce a
structure-based alignment and SWISS-MODEL was used in the optimized
mode to minimize energy (14,16).
For structural evaluation and stereochemical analyses, the 3D model
was submitted to the EBI PDBsum pictorial database (17). The M. tuberculosis CysE
protein model was further evaluated using the University of
California, Los Angeles-Department of Energy Institute programs
PROCHECK (18), Verify3D and ERRAT
(http://services.mbi.ucla.edu).
Site-directed mutagenesis of M.
tuberculosis cysE
Three pairs of mutated primers were designed
according to the DNA sequence of M. tuberculosis cysE
(Table I). Every pair mutated
primer was characterized with their 5′ends adjacent and 3′ends in
opposite directions according to the instructions of the Takara
MutanBEST kit (Takara Bio, Inc.). The recombinant plasmid
pMD18-Mtb-cysE, which contains NdeI and XhoI
restriction sites, was constructed previously (7) and was used as the template. The
linear mutated plasmids were amplified using Pfu DNA
polymerase (Thermo Fisher Scientific, Waltham, MA, USA). The
amplification reaction was performed in a final volume of 50 µl
mixture including 0.5 µl Pfu DNA polymerase (5 U/µl), 5 µl
Pfu Buffer (10X), 5 µl dNTP mixture (2.5 mM each), 1 µl
template DNA (100 ng), 3 µl mutated primers (20 µM) and
ddH2O. The reaction conditions consisted of
pre-denaturation (94°C, 5 min), 30 cycles of amplification
(denaturation at 94°C for 30 sec, annealing at 65/61°C for 30 sec
and primer extension at 72°C for 5 min) and final extension at 72°C
for 10 min. Plasmids were recircularized by blunting kination
ligation and self-ligation using Takara MutanBEST kit (Takara Bio,
Inc.). pMD18-Mtb-cysE plasmids containing site-directed mutations
were transformed and grown in E. coli Novablue competent
cells (Merck Millipore). The plasmid was transformed into competent
cells by heat shock at 42°C. The E. coli Novablue cells
harboring plasmid were cultured in lysogeny medium supplemented
with 100 ug/ml ampicillin (Merck Millipore) at 37°C. Positive
clones were identified by restriction endonuclease digestion
(Takara Bio, Inc.) and DNA sequencing as those harboring the
required mutations. The sequencing work was collaborated with
Takara Bio Inc.
 | Table I.Sequences of oligonucleotide primers
used for constructing site-direct mutations of M. tuberculosis
cysE gene. |
Table I.
Sequences of oligonucleotide primers
used for constructing site-direct mutations of M. tuberculosis
cysE gene.
| Name | Primer
orientation | Primer
sequencea
(5′-3′) |
|---|
| D67A | F |
ATCCTGACCGGTGTAGCCATCC |
|
|
| ACCCCGGTGCC |
|
| R |
TACACCGGTCAGGATGCG |
| H82A | F |
CGCGTGTTCATCGACGCCGCG |
|
|
| ACCGGCGTGGTG |
|
| R |
GTCGATGAACACGCGAGCACCG |
| H117A | F |
GTTGGCGGGAAACGCGCCCCC |
|
|
| ACCGTCGGTGAC |
|
| R |
GCGTTTCCCGCCAACCATGCC |
Preparation of mutant M. tuberculosis
CysE proteins
Site-mutated M. tuberculosis cysE genes were
subcloned into pET29b (Merck Millipore) at NdeI and
XhoI restriction sites, and transformed into E. coli
BL21 (DE3) cells (Merck Millipore) to express the three different
CysE mutated proteins using the heat shock method, each containing
a polyhistidine (His)-tag. CysE mutant proteins were prepared and
purified using Ni2+ affinity chromatography according to the
expression protocol of wild-type M. tuberculosis CysE
protein, as previously described (7). Total protein (~10 mg) was loaded onto
a 1 ml Ni-NTA agarose column (Qiagen, Hilden, Germany). Then the
column was washed with 20 ml wash buffer (20 mM Tris-HCl pH 8.0,
500 mM NaCl, 20% glycerol, 60 mM imidazole and 1 mM PMSF). Finally,
the mutant CysE proteins with a His-tag at the C-terminus were
eluted with 10 ml elution buffer (20 mM Tris-HCl pH 8.0, 500 mM
NaCl, 20% glycerol, 300 mM imidazole and 1 mM PMSF). The
concentration of purified mutant CysE proteins was measured using
the BCA Protein Assay Kit (P0012S; Beyotime Institute of
Biotechnology, Haimen, China). The purified mutant CysE proteins
were identified by western blot analysis. The protocol was as
followed: Purified proteins (20 µg) per lane were loaded and
sepearated by 12% SDS-PAGE (P0012A; Beyotime Institute of
Biotechnology). The proteins were transferred to nitrocellulose
membranes (Thermo Fisher Scientific, Inc.) after electrophoresed
with SDS-PAGE. Following blocking in 5% skimmed milk buffer for 2 h
at 37°C, the membrane was incubated with mouse antipolyhistidine
monoclonal antibody (dilution, 1:5,000; H1029; Merck Millipore) for
2 h at 37°C and followed by horse-anti-mouse IgG conjugated with
alkaline phosphatase (dilution, 1:2,000; ZB-2310; ZSGB-Bio,
Beijing, China) for 45 min at room temperature. After staining in
the solution mixture of BCIP/NBT (ZSGB-Bio), the immunoreactive
protein bands were visualized and collected.
Enzymatic activity assay for mutant
CysE proteins
The enzymatic activity assay of the various CysE
mutants was conducted by the Ellman's reagent
[5,5′-dithiobis-(2-nitrobenzoic acid); DTNB] method, as previously
described (19). Briefly, equal
concentrations of the purified CysE mutant proteins and the
wild-type M. tuberculosis CysE protein were used. The
relative activities of the mutated CysE proteins were calculated
against activity of the wild-type M. tuberculosis CysE,
which was used as the control (set to 100%). All data were analyzed
using GraphPad Prism 6 (GraphPad, La Jolla, CA, USA).
Results and Discussion
Sequence analysis and alignment
ProtParam was used to analyze various physiochemical
properties from the amino acid sequence of CysE protein. The
molecular weight and pI of the 229-amino-acid-long sequence were
predicted as 23,770.3 Da and pH 7.11, respectively. An instability
index of 27.56 indicated that the CysE protein was stable. The
GRAVY index of 0.217 was indicative of a hydrophobic protein.
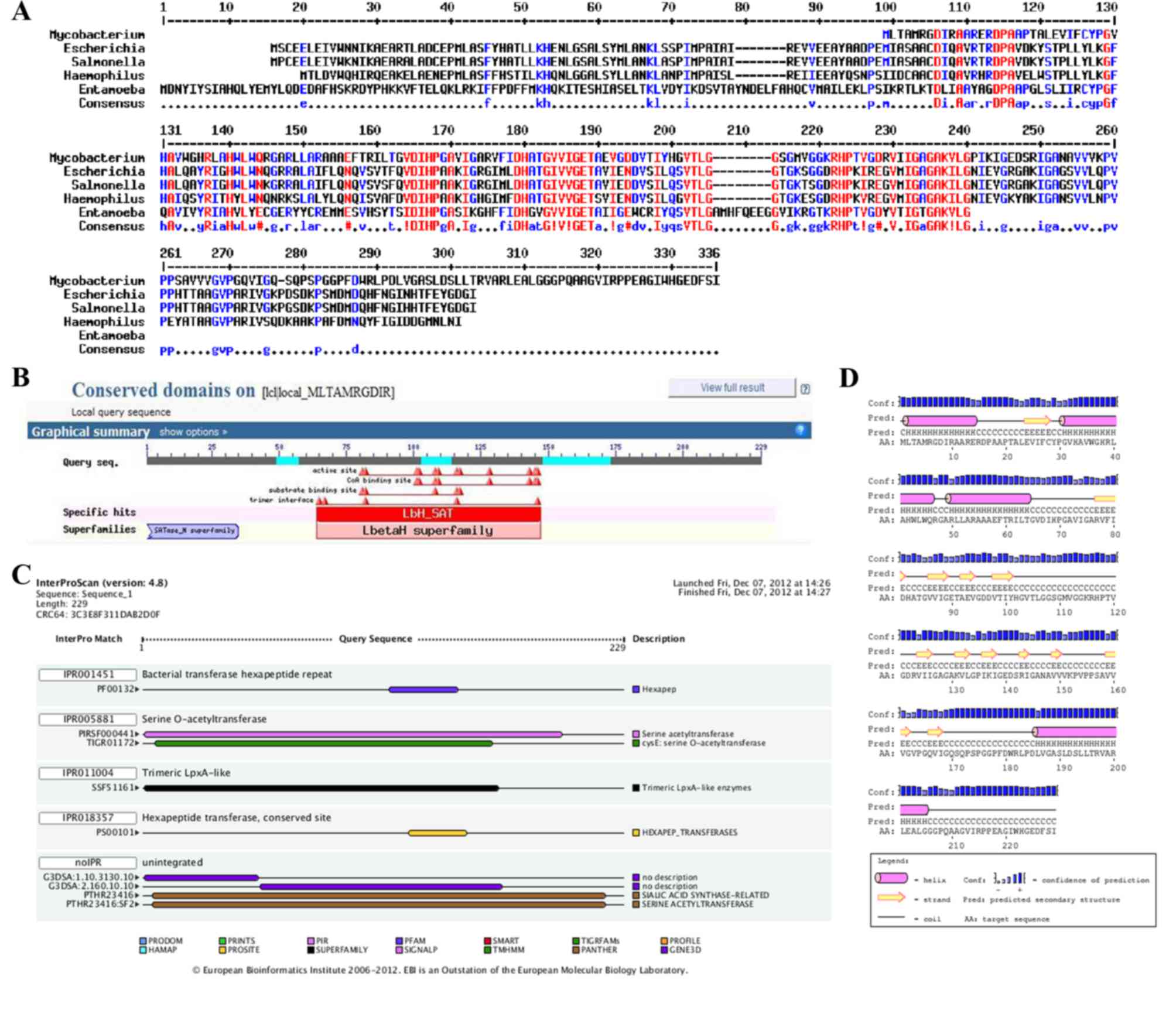
Sequence alignment demonstrated that the M. tuberculosis
CysE protein shared identity with E. coli CysE (45%), S.
typhimurium CysE (45%), H. influenzae CysE (45%) and
E. histolytica CysE (49%) (Fig.
1A).
Secondary structure prediction and
functional annotation of CysE protein
The bioinformatics tools InterProScan, PSIPRED and
NCBI-CDD were used to search the conserved domains and potential
functions of the M. tuberculosis CysE protein. The results
revealed that the CysE protein possessed a left-handed-β-helix
(LβH) domain and belonged to the hexapeptide acetyltransferase
family (Fig. 1B-D), as previously
shown for Rhizobium leguminosarm (20). This family of acetyltransferases is
characterized by imperfect tandem repeats of the hexapeptide motif
[LIV]-[GAED]-X2-[STAV]-X (21)
with a conserved LβH domain, which is folded by three hexapeptides
(22,23).
3D modeling of the CysE protein
Homology modeling is a standard method for structure
prediction and contributes to the understanding of the relationship
between protein structure and function. The SWISS-MODEL server was
used to model proteins based on protein-structure homology
(24). A PSI-BLAST search revealed
3P47A as an available template that can be used for homology
modeling of the CysE protein. 3P47A is a refined X-ray diffraction
model of the A-chain of E. histolytica CysE at a resolution
of 1.78 Å. Homology modeling of M. tuberculosis CysE was
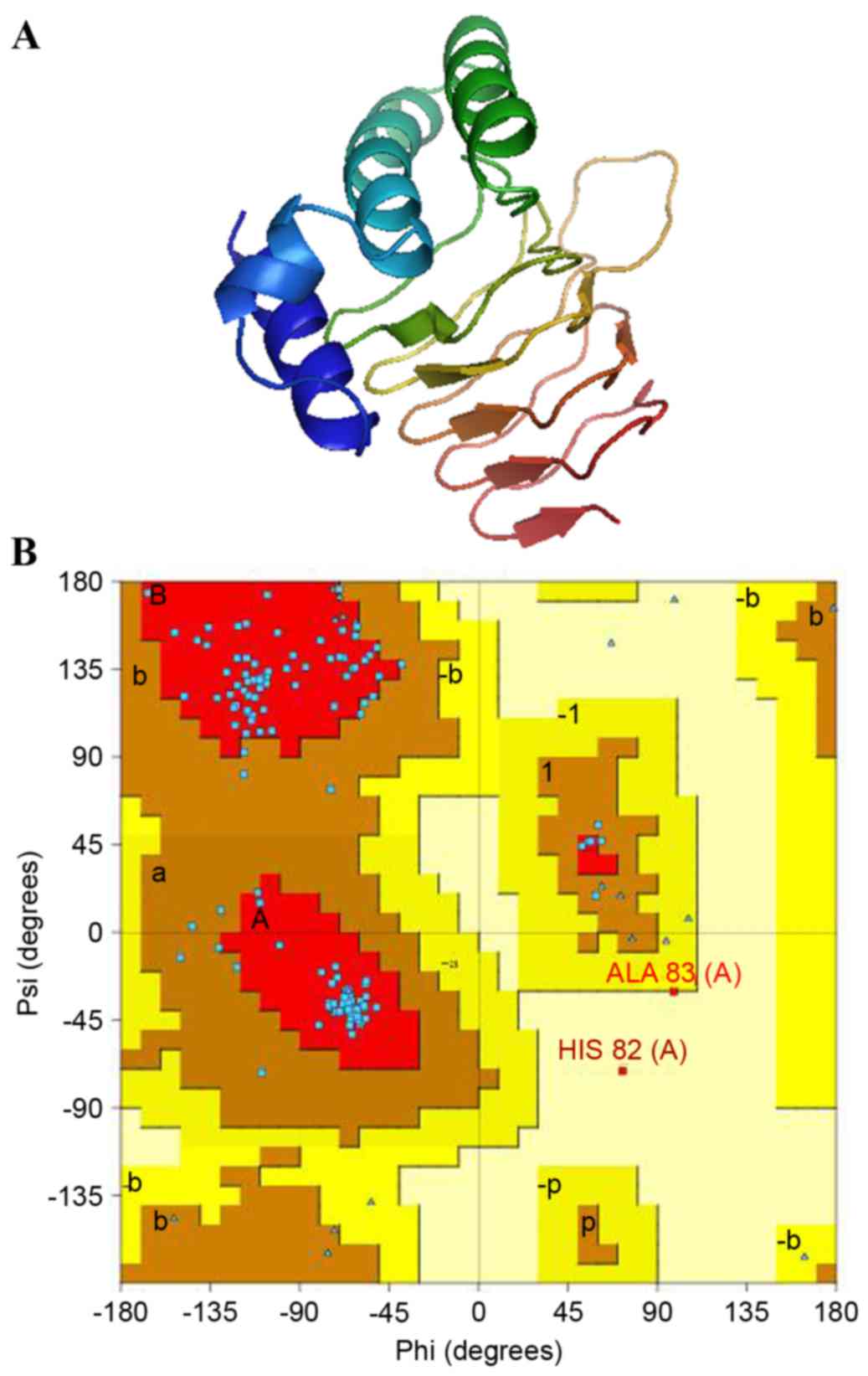
constructed using 3P47A as a template (Fig. 2A).
Energy minimization, quality
assessment and visualization
The reliability of the predicted 3D model of the
M. tuberculosis CysE was assessed by Ramachandran plot,
using PROCHECK, and a quality assessment confirmed using PDBsum.
The Ramachandran plot revealed a distribution of 160 amino acid
residues of M. tuberculosis CysE, with the majority of
residues (98.4%) in the favored and allowed regions, and only two
of the residues (1.6%) in the generously allowed region (Fig. 2B). The g-factor demonstrates how
unusual a stereochemical property is: Values below −0.5 represent
unusual property, whereas values below −1.0 indicate high
unusualness. The g-factors for dihedral angles and main chain
covalent forces were −0.10 and 0.31, respectively. The overall
average g-factor for the M. tuberculosis CysE model was
0.07. Taken together, the Ramachandran plot and g-factors indicated
that the backbone dihedral angles, phi and psi in the 3D model of
M. tuberculosis CysE were within acceptable limits.
Following the construction of a predicted 3D
structure, the final protein model was verified using Verify3D,
which analyzed the compatibility of the 3D (atomic) model of CysE
with its own amino acid sequence (1D). The scores of a sliding
21-residue window (between −10 and +10) were added and plotted for
the individual residues (25,26).
The M. tuberculosis CysE protein was consistent with the
respective amino acids, with 88.82% of the residues having an
averaged 3D-1D and a score >0.2. ERRAT is a protein structure
verification algorithm that is well suited for evaluating the
quality of crystallographic model building and refinement. The
final model had a quality factor of 91.156% in ERRAT, indicating
that the model was acceptable and credible.
Site-directed mutagenesis of M.
tuberculosis CysE
The conserved residues D67, H82 and H117 of M.
tuberculosis CysE were selected for site-directed mutation. To
maintain the native spatial structure, these three residues were
substituted with alanine. Three linear pMD18 plasmid vectors
containing the mutated M. tuberculosis cysE gene
(pMD18-Mtb-cysE) were amplified using mutated primers. Restriction
endonuclease digestion and sequencing analysis confirmed that the
three specific site-directed mutations were obtained successfully
(data not shown).
Preparation of mutant M. tuberculosis
CysE proteins
The pET29b-Mtb-cysE expression plasmids containing
the subcloned site-directed mutations were constructed and
transformed into E. coli. Three different soluble CysE
mutant proteins with fused His-tags were expressed in E.
coli BL21 (DE3) cells, and purified by Ni2+ affinity
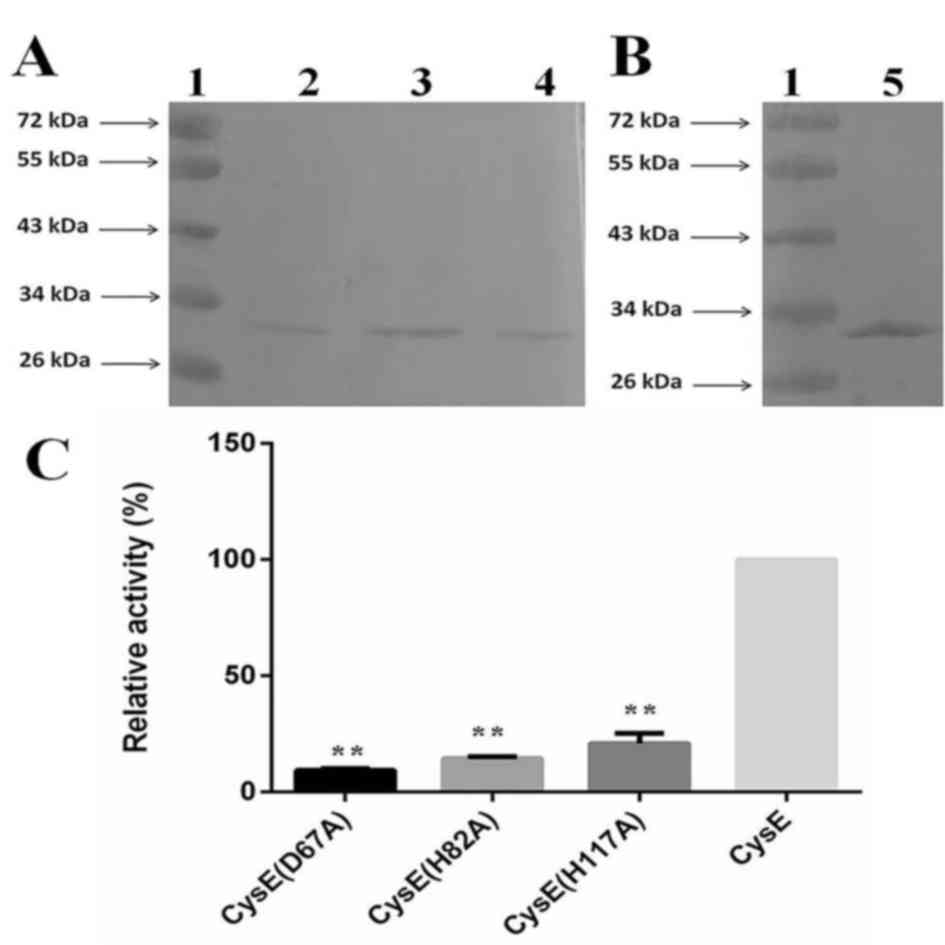
chromatography. The purified mutant proteins were identified by
western blot analysis. The band of CysE mutant proteins appeared at
~30 kDa, which was similar to that of wild-type M.
tuberculosis CysE protein (7)
(Fig. 3A and B).
Enzymatic activity assay for mutant
CysE proteins
CysE enzymatic activity of the three mutant proteins
was detected using a colorimetric assay with DTNB (19), which indicated that the mutant
M. tuberculosis CysE proteins exhibited different enzymatic
activity compared with wild-type CysE. The D67A mutant demonstrated
only 9.79% activity, whereas the H82A and H117A mutants had 14.31
and 20.82% activity, respectively (Fig. 3C). These data suggested that the
three residues may be involved in the functional activity of the
enzyme.
In conclusion, site-directed mutagenesis of CysE and
the enzymatic activity assay indicated that the conserved amino
acid residues D67, H82 and H117 may be involved in the catalytic
activity and, thus, the function of CysE. The prediction for M.
tuberculosis CysE structure may facilitate the elucidation of
the functional mechanism of this enzyme. However, further studies
regarding the interaction between Cys as a substrate and M.
tuberculosis CysE as an enzyme are required. Furthermore,
determination of the active amino acids of M. tuberculosis
CysE will immensely promote the development of structure-guided
enzyme inhibitors for use as novel and effective anti-TB drugs.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 31070066).
References
|
1
|
Cole ST and Riccardi G: New tuberculosis
drugs on the horizon. Curr Opin Microbiol. 14:570–576. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hell R: Molecular physiology of plant
sulfur metabolism. Planta. 202:138–148. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kredich NM, Becker MA and Tomkins GM:
Purification and characterization of cysteine synthetase, a
bifunctional protein complex, from Salmonella typhimurium. J Biol
Chem. 244:2428–2439. 1969.PubMed/NCBI
|
|
4
|
Kredich NM and Tomkins GM: The enzymic
synthesis of L-cysteine in Escherichia coli and Salmonella
typhimurium. J Biol Chem. 241:4955–4965. 1966.PubMed/NCBI
|
|
5
|
Schnell R and Schneider G: Structural
enzymology of sulphur metabolism in Mycobacterium tuberculosis.
Biochem Biophys Res Commun. 396:33–38. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Raman K, Yeturu K and Chandra N: targetTB:
A target identification pipeline for Mycobacterium tuberculosis
through an interactome, reactome and genome-scale structural
analysis. BMC Syst Biol. 2:1092008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qiu J, Wang D, Ma Y, Jiang T and Xin Y:
Identification and characterization of serine acetyltransferase
encoded by the Mycobacterium tuberculosis Rv2335 gene. Int J Mol
Med. 31:1229–1233. 2013.PubMed/NCBI
|
|
8
|
Benson DA, Karsch-Mizrachi I, Lipman DJ,
Ostell J and Wheeler DL: GenBank. Nucleic Acids Res. 36:(Database
Issue). D25–D30. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wilkins MR, Gasteiger E, Bairoch A,
Sanchez JC, Williams KL, Appel RD and Hochstrasser DF: Protein
identification and analysis tools in the ExPASy server. Methods Mol
Biol. 112:531–552. 1999.PubMed/NCBI
|
|
10
|
Corpet F: Multiple sequence alignment with
hierarchical clustering. Nucleic Acids Res. 16:10881–10890. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zdobnov EM and Apweiler R: InterProScan-an
integration platform for the signature-recognition methods in
InterPro. Bioinformatics. 17:847–848. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
McGuffin LJ, Bryson K and Jones DT: The
PSIPRED protein structure prediction server. Bioinformatics.
16:404–405. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marchler-Bauer A, Derbyshire MK, Gonzales
NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI,
et al: CDD: NCBI's conserved domain database. Nucleic Acids Res.
43:(Database Issue). D222–D226. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guex N, Peitsch MC and Schwede T:
Automated comparative protein structure modeling with SWISS-MODEL
and Swiss-PdbViewer: A historical perspective. Electrophoresis.
30:(Suppl 1). S162–S173. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Altschul SF, Madden TL, Schäffer AA, Zhang
J, Zhang Z, Miller W and Lipman DJ: Gapped BLAST and PSI-BLAST: A
new generation of protein database search programs. Nucleic Acids
Res. 25:3389–3402. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guex N and Peitsch MC: SWISS-MODEL and the
Swiss-PdbViewer: An environment for comparative protein modeling.
Electrophoresis. 18:2714–2723. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Laskowski RA: PDBsum: Summaries and
analyses of PDB structures. Nucleic acids Res. 29:221–222. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Laskowski RA, Rullmannn JA, MacArthur MW,
Kaptein R and Thornton JM: AQUA and PROCHECK-NMR: Programs for
checking the quality of protein structures solved by NMR. J Biomol
NMR. 8:477–486. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Riddles PW, Blakeley RL and Zerner B:
Reassessment of Ellman's reagent. Methods Enzymol. 91:49–60. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Downie JA: The nodL gene from Rhizobium
leguminosarum is homologous to the acetyl transferases encoded by
lacA and cysE. Mol Microbiol. 3:1649–1651. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vaara M: Eight bacterial proteins,
including UDP-N-acetylglucosamine acyltransferase (LpxA) and three
other transferases of Escherichia coli, consist of a six-residue
periodicity theme. FEMS Microbiol Lett. 76:249–254. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jenkins J and Pickersgill R: The
architecture of parallel beta-helices and related folds. Prog
Biophys Mol Biol. 77:111–175. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Raetz CR and Roderick SL: A left-handed
parallel beta helix in the structure of UDP-N-acetylglucosamine
acyltransferase. Science. 270:997–1000. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Arnold K, Bordoli L, Kopp J and Schwede T:
The SWISS-MODEL workspace: A web-based environment for protein
structure homology modelling. Bioinformatics. 22:195–201. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Luthy R, Bowie JU and Eisenberg D:
Assessment of protein models with three-dimensional profiles.
Nature. 356:83–85. 1992. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bowie JU, Luthy R and Eisenberg D: A
method to identify protein sequences that fold into a known
three-dimensional structure. Science. 253:164–170. 1991. View Article : Google Scholar : PubMed/NCBI
|