Introduction
Emanuel syndrome (ES), also referred to as
derivative 22 syndrome, derivative 11;22 syndrome, partial trisomy
11;22, or supernumerary der(22)
t(11;22) syndrome (1,2), is an unbalanced translocation
syndrome that usually results from a 3:1 meiotic disjunction of a
parental balanced translocation between chromosomes 11 and 22
during gametogenesis. In >99% of patients with ES, one of the
parents is a balanced carrier of a t(11;22)(q23;q11.2)
translocation and normal phenotype. The possible outcomes of future
pregnancies of the parents include: Normal chromosomes, Emanuel
syndrome, balanced t(11;22) carrier, and spontaneous abortion as a
result of ES or another meiotic malsegregation. ES is characterized
by severe intellectual disability, microcephaly, failure to thrive,
preauricular tag or sinus, ear anomalies, cleft or high-arched
palate, micrognathia, congenital heart disease, anal atresia,
diaphragmatic hernia and genital abnormalities in men (3). The prevalence of ES is estimated at 1
in 110,000, and long-term survival is possible (4,5).
Referring to the treatment strategies of ES, care provided by a
multidisciplinary team is essential, and includes standard
management of anal atresia, diaphragmatic hernia, cardiac defects,
hearing loss, cleft palate and speech therapies.
Clinical diagnosis of ES depends on the distinct
phenotype above. The molecular genetic testing foe ES consists of
chromosome analysis, fluorescence in situ hybridization,
chromosomal microarray analysis. In the present study, chromosome
analysis and chromosomal microarray analysis, which usually include
comparative genetic hybridization and single nucleotide
polymorphism (SNP)-arrays, were performed. Chromosomal microarray
analysis has greatly improved the available chromosome resolution
and the identification of chromosomal abnormalities, particularly
in defining the breakpoint of rearrangements and the accurate
detection of copy number variants. The present study aimed to
confirm the first patient with ES in China, and to promote the
understanding of this syndrome in mainland China.
Materials and methods
Ethical approval and patient
consent
This study was approved by the Review Board of The
Second Xiangya Hospital of Central South University (Changsha,
China). Written informed consent was obtained from the parents of
the patient for publication of this study and any accompanying
images.
Clinical presentation
The proband, a 2-year-old boy, was born full term to
a 26-year-old G1P1 mother and a 30-year-old father. The family
history is unremarkable. The mother had a history of threatened
abortion at 8 weeks gestation and underwent tocolytic treatment at
a local hospital. At 5 months old, the patient received surgery to
repair a cleft lip; at this time, a cardiac murmur was identified
and echocardiography revealed an atrial septal defect (ASD). The
child was referred to The Second Xiangya Hospital of Central South
University (Changsha, China) and the ASD was repaired in December
2014; however, during surgery, thymic dysplasia was revealed.
Karyotype analysis determined the proband to be 47,XY,+del
(22)(q13) and the maternal
karyotype was 46,XX,t(11;22)(q25;q13),9qh-,15p+. During
hospitalization, it was noted that the patient was unable to speak
and walk. The Gesell Maturation Scale determined a developmental
quotient of 33, which indicates an intelligence level equal to a
30-week-old infant, and is attributed to the severe mental and
developmental retardation of the patient. Hearing impairment was
detected using an auditory acuity test. Physical examination
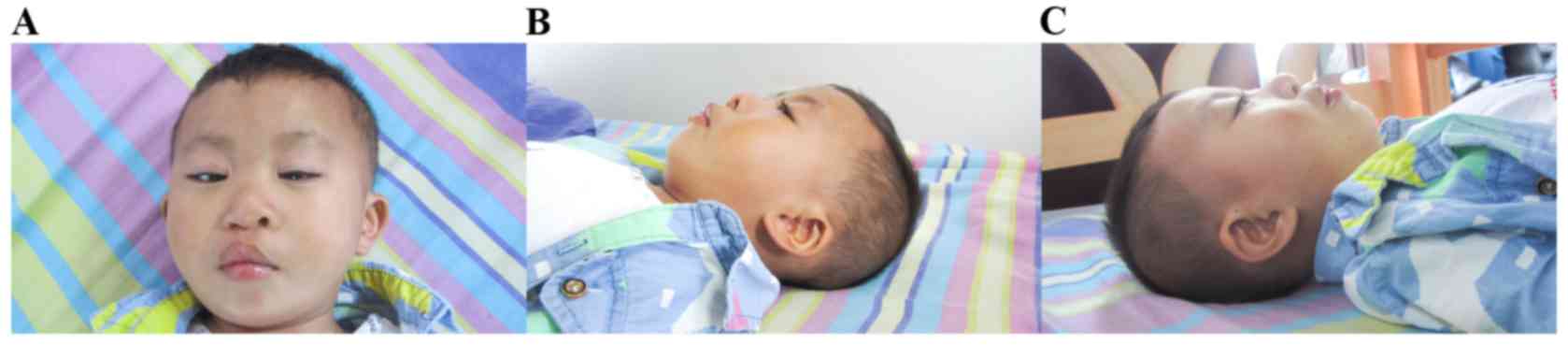
revealed facial dysmorphism, cleft lip and palate, genital
malformation (micropenis) and amblyopia. Dysmorphic features
included a prominent forehead, widely separated eyes with
down-slanting palpebral fissure, broad nasal bridge, prominent
philtrum, and bilateral large and low-set ears (Fig. 1).
Follow-up was completed in September 2015; at this
time echocardiography revealed no left-to-right shunt at the atrial
septum, and the ejection fraction was normal.
Cytogenetic analysis
Peripheral blood (5 ml) was collected from the
patient and each of the parents, and chromosome analysis was
performed by conventional G-banding techniques (550-band
resolution). All samples were subjected to lymphocyte culture
according to standard cytogenetic protocol (6).
SNP-array analysis
Genomic DNA was isolated from peripheral blood of
the patient and the two parents using the DNeasy Blood & Tissue
kit (Qiagen, Inc., Valencia, CA, USA) with the QIAcube automated
DNA extraction robot (Qiagen GmbH, Hilden, Germany). Genomic DNA
samples were adjusted to a final concentration of 50 ng/ml. The
HumanOmni1-Quad BeadChip (Illumina, Inc., San Diego, CA, USA) and
the Illumina BeadScan genotyping system (Beadstation Scanner;
Illumina, Inc.) were employed to obtain the signal intensities of
the SNP probes (7–9). The HumanOmni1-Quad Beadchip contains
>1.1 million loci across the human genome, including markers
derived from the International HapMap Project, the 1000 Genomes
Project (http://www.internationalgenome.org/) and previously
published studies (10). The human
genome assembly GRCh37/hg19 from the University of California,
Santa Cruz Genome Browser (Santa Cruz, CA, USA) was used with
Illumina GenomeStudio v2011 (Illumina, Inc.) software to analyze
the genotypes and to evaluate the experimental quality. The call
rates of the samples are >99.2%.
Results
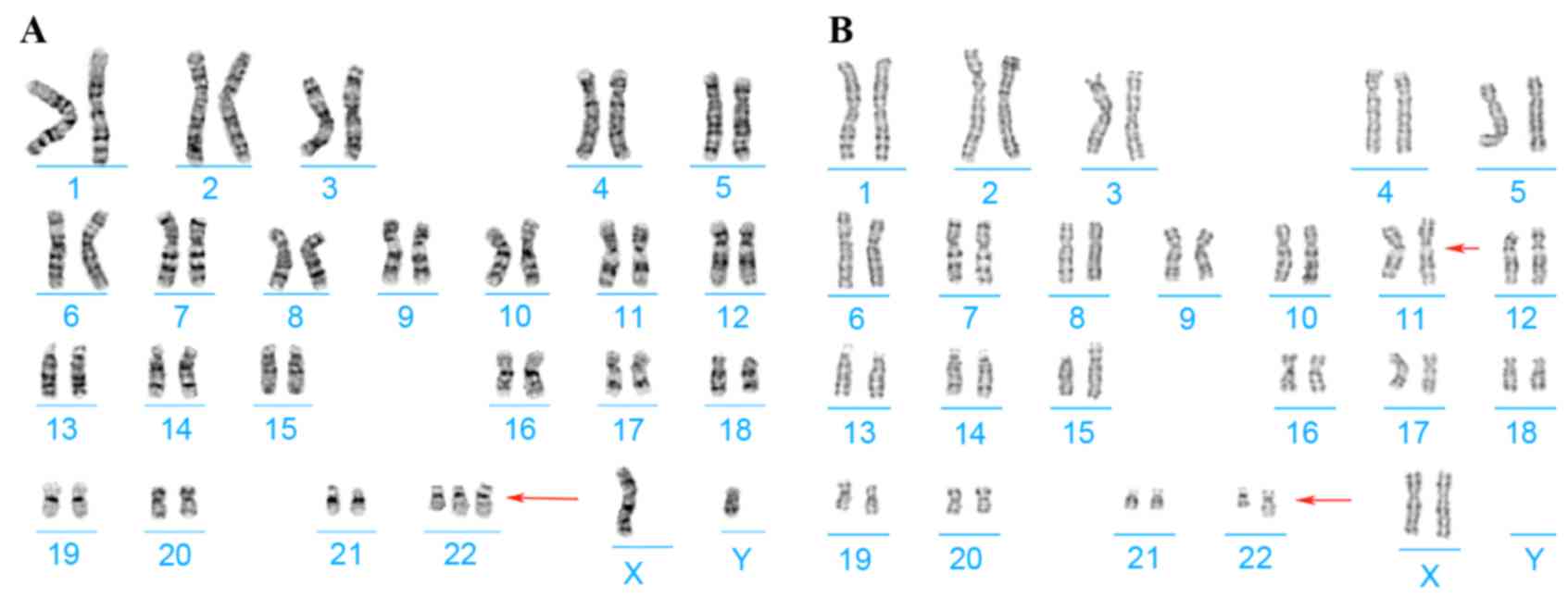
Chromosome analysis of the proband and his parents
was performed using G-banding techniques on stimulated blood
lymphocytes. Cytogenetics revealed the karyotypes of the proband
and his mother to be 47,XY,+del(22)(q13) and
46,XX,t(11;22)(q25;q13),9qh-,15p+, respectively (Fig. 2); his father had a normal karyotype
result.
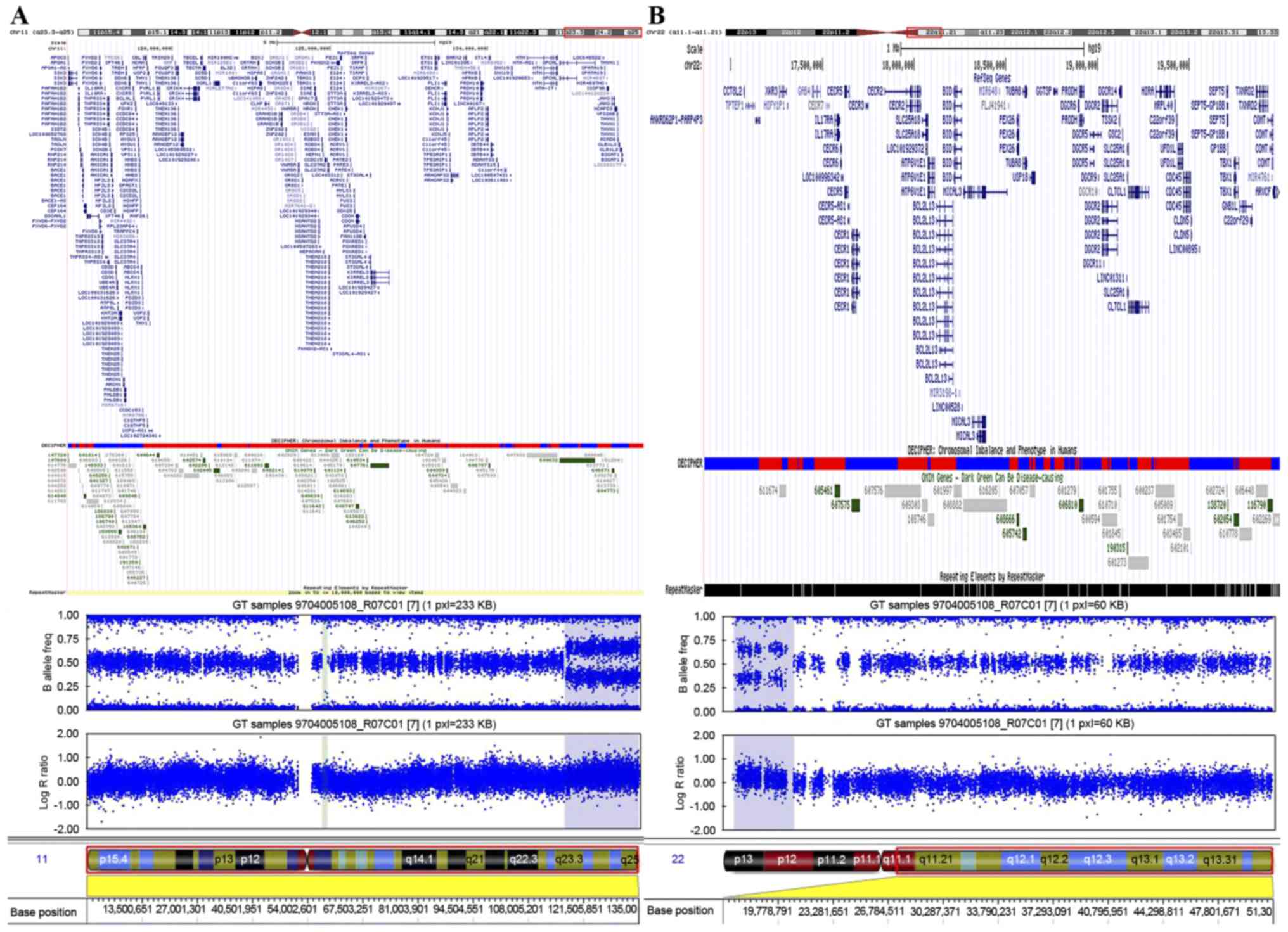
To define the chromosomal abnormality, SNP-array
analysis was performed on the proband's genomic DNA using the
Illumina HumanOmni1-Quad array (Fig.
3). This analysis revealed a region of gain that spanned ~3.1
Mb on chromosome 22q11.1-q11.21, between 16,855,618 and 19,995,480
bp (Fig. 3B). In addition to the
aforementioned duplication, the present study detected a
duplication of ~18.2 Mb in chromosome region 11q23.3-q25, between
116,696,681 and 134,942,926 bp (Fig.
3A). The diagnosis of ES was confirmed by comparing the
breakpoints identified in this patient with those reported in other
cases of ES (11). Maternal
SNP-array analysis was normal.
Discussion
ES is an inherited chromosomal abnormality syndrome
(3) that consists of a derivative
chromosome 22 as a supernumerary chromosome with a karyotype of
47,XX,+der(22)t(11;22)(q23;q11)
in females or 47,XY,+der(22)t(11;22)(q23;q11) in males (5). In 2004, this chromosomal imbalance
was named as ES (OMIM no. 609029) (1,3)
According to previous reports, >100 individuals with ES have
been reported (1,3,12–16);
however, the patient in the present study is the first, to the best
of our knowledge, to be reported in China.
Patients with ES exhibit a distinctive phenotype,
and global developmental delay is observed in almost all patients.
Congenital heart disease is present in ~57% subjects, with the
three most common anomalies being ASD, ventricular septal defect
and patent ductus arteriosus. Other heart malformations, including
tetralogy of Fallot, coarctation of the aorta, pulmonic stenosis,
total anomalous pulmonary venous connection, truncus arteriosus,
transposition of the great arteries and tricuspid atresia, have
previously been reported (1,5,16–18).
In the present study, besides ASD, the proband presented with
severe intellectual disability and developmental retardation,
congenital heart disease, facial dysmorphism, cleft lip and palate,
thymic dysplasia, hearing impairment, genital malformation
(micropenis) and amblyopia; these clinical features have been
observed in other patients with ES.
Based on the clinical features of the proband and
the karyotype of his mother, the diagnosis of ES was considered,
but not confirmed, following karyotype analysis.
Translocation-specific polymerase chain reaction (PCR) is the most
cost-effective diagnostic method for der(22)t(11;22) and der (22)t(8;22) (19); however, other chromosomal
abnormalities cannot be detected using this method. For this
reason, the more accurate and precise SNP-array is used to verify
the results of translocation-specific PCR. Therefore, SNP-array
analysis was considered to further investigate the genetic
variation of the proband. SNP-array analysis identified two
pathogenic duplications, which involved 22q11.1-q11.21 (3.1Mb) and
11q23.3-q25 (18.2Mb), respectively. This result confirmed the
diagnosis of ES and demonstrated the limit of karyotype analysis,
suggesting that a high-resolution technique, such as the SNP-array
or array comparative genomic hybridization, should be recommended
when karyotype analysis is uncertain.
Duplication of 22q11.2 has been reported to present
a highly variable phenotype that ranges from healthy individuals to
those with learning disabilities and congenital defects (20–23).
Some of the main clinical features of 22q11.2 duplication,
including facial dysmorphism, hearing impairment and congenital
heart disease, were present in the patient in the present study.
One crucial gene that is affected by 22q11.2 duplication is T-box
1, which contributes to the phenotype of the proband with ES.
Duplication of 11q23.3-q25 without a copy number
change of another chromosome is rare (24–28)
and shares several common clinical features with 22q11.2
duplication, such as intellectual disability, growth retardation,
microcephaly, facial dysmorphism, epilepsy, congenital inguinal
hernia, and cardiac, renal and cerebral malformations (27,29),
some of which were presented in the patient in the present study.
The duplication region of this patient contains >100 OMIM genes,
many of which have not been well characterized. The genes that
contribute to the phenotypes of 11q23.3-q25 duplication and the
mechanisms by which changes in gene dosage exert disruptive effects
on gene structure and function remain unknown. Above all, the
clinical phenotype of ES arises from the duplication of 22q11 and
11q23-qter (25).
When the mother is a balanced translocation carrier,
there is an increased sibling recurrence-risk ratio of children
with ES, as well as pregnancy loss due to other types of unbalanced
translocations. Genetic counseling is essential and indispensable
for this population. Furthermore, prenatal diagnosis could be
performed through chorionic villus biopsy or amniocentesis.
In conclusion, the present study described the
clinical features of a patient with ES. Although >100 patients
with ES have been reported worldwide, the proband in the present
study is the first, to the best of our knowledge, to be reported in
China. Considering the prevalence of ES and the population of
China, there are likely to be numerous patients with undiagnosed
ES. The present study emphasizes the necessity and importance of
high-resolution techniques for the molecular diagnosis of
chromosomal abnormalities, and contributes to the further mapping
and phenotypic delineation of ES.
Acknowledgements
The authors of the present study would like to thank
the State Key Laboratory of Medical Genetics of China for technical
assistance. This study was supported by the National Natural
Science Foundation of China (grant no. 81500245 to LX, grant no.
81101475 to ZPT, and grant no. 81370204 to YFY).
References
|
1
|
Fraccaro M, Lindsten J, Ford CE and
Iselius L: The 11q;22q translocation: A European collaborative
analysis of 43 cases. Hum Genet. 56:21–51. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hou JW: Supernumerary chromosome marker
Der(22)t(11;22) resulting from a maternal balanced translocation.
Chang Gung Med J. 26:48–52. 2003.PubMed/NCBI
|
|
3
|
Zackai EH and Emanuel BS: Site-specific
reciprocal translocation, t(11;22)(q23;q11), in several unrelated
families with 3:1 meiotic disjunction. Am J Med Genet. 7:507–521.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ohye T, Inagaki H, Kato T, Tsutsumi M and
Kurahashi H: Prevalence of Emanuel syndrome: Theoretical frequency
and surveillance result. Pediatr Int. 56:462–466. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Carter MT, St Pierre SA, Zackai EH,
Emanuel BS and Boycott KM: Phenotypic delineation of Emanuel
syndrome (supernumerary derivative 22 syndrome): Clinical features
of 63 individuals. Am J Med Genet A. 149A:1712–1721. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Benn P and Delach J: Human lymphocyte
culture and chromosome analysis. CSH Protoc. 2008:pdb. prot5035.
2008.PubMed/NCBI
|
|
7
|
Xie L, Chen JL, Zhang WZ, Wang SZ, Zhao
TL, Huang C, Wang J, Yang JF, Yang YF and Tan ZP: Rare de novo copy
number variants in patients with congenital pulmonary atresia. PLoS
One. 9:e964712014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tang M, Yang YF, Xie L, Chen JL, Zhang WZ,
Wang J, Zhao TL, Yang JF and Tan ZP: Duplication of 10q22.3-q23.3
encompassing BMPR1A and NGR3 associated with congenital heart
disease, microcephaly, and mild intellectual disability. Am J Med
Genet A. 167A:3174–3179. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sun G, Tan Z, Fan L, Wang J, Yang Y and
Zhang W: 1q21.1 microduplication in a patient with mental
impairment and congenital heart defect. Mol Med Rep. 12:5655–5658.
2015.PubMed/NCBI
|
|
10
|
Pinto D, Darvishi K, Shi X, Rajan D,
Rigler D, Fitzgerald T, Lionel AC, Thiruvahindrapuram B, Macdonald
JR, Mills R, et al: Comprehensive assessment of array-based
platforms and calling algorithms for detection of copy number
variants. Nat Biotechnol. 29:512–520. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kurahashi H, Shaikh TH, Zackai EH, Celle
L, Driscoll DA, Budarf ML and Emanuel BS: Tightly clustered 11q23
and 22q11 breakpoints permit PCR-based detection of the recurrent
constitutional t(11;22). Am J Hum Genet. 67:763–768. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Biederman BM, Lin CC, Lowry RB and
Somerville R: Tertiary trisomy (22q11q),47,+der(22),t(11;22). Hum
Genet. 53:173–177. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pihko H, Therman E and Uchida IA: Partial
11q trisomy syndrome. Hum Genet. 58:129–134. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schinzel A, Schmid W, der Auf Maur P,
Moser H, Degenhardt KH, Geisler M and Grubisic A: Incomplete
trisomy 22. I. Familial 11/22 translocation with 3:1 meiotic
disjunction. Delineation of a common clinical picture and report of
nine new cases from six families. Hum Genet. 56:249–262. 1981.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Iselius L, Lindsten J, Aurias A, Fraccaro
M, Bastard C, Bottelli AM, Bui TH, Caufin D, Dalprà L and Delendi
N: The 11q;22q translocation: A collaborative study of 20 new cases
and analysis of 110 families. Hum Genet. 64:343–355. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin AE, Bernar J, Chin AJ, Sparkes RS,
Emanuel BS and Zackai EH: Congenital heart disease in supernumerary
der(22), t(11;22) syndrome. Clin Genet. 29:269–275. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Giraud F, Mattei JF, Mattei MG and Bernard
R: Partial trisomy 11q and familial translocation 11–22 (author's
transl). Humangenetik. 28:343–347. 1975.(In French). PubMed/NCBI
|
|
18
|
Pangalos C, Couturier J, Bartsocas C and
Theodorou S: Partial 11q trisomy due to missegregation of maternal
t(11;22) (q23;q11.1) translocation (author's transl). Nouv Presse
Med. 9:3065–3067. 1980.(In French). PubMed/NCBI
|
|
19
|
Mishra D, Kato T, Inagaki H, Kosho T,
Wakui K, Kido Y, Sakazume S, Taniguchi-Ikeda M, Morisada N, Iijima
K, et al: Breakpoint analysis of the recurrent constitutional
t(8;22) (q24.13;q11.21) translocation. Mol Cytogenet. 7:552014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Alberti A, Romano C, Falco M, Calì F,
Schinocca P, Galesi O, Spalletta A, Di Benedetto D and Fichera M:
1.5 Mb de novo 22q11.21 microduplication in a patient with
cognitive deficits and dysmorphic facial features. Clin Genet.
71:177–182. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Courtens W, Schramme I and Laridon A:
Microduplication 22q11.2: A benign polymorphism or a syndrome with
a very large clinical variability and reduced penetrance?-Report of
two families. Am J Med Genet A. 146A:758–763. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ensenauer RE, Adeyinka A, Flynn HC,
Michels VV, Lindor NM, Dawson DB, Thorland EC, Lorentz CP,
Goldstein JL, McDonald MT, et al: Microduplication 22q11.2, an
emerging syndrome: Clinical, cytogenetic, and molecular analysis of
thirteen patients. Am J Hum Genet. 73:1027–1040. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lundin J, Söderhäll C, Lundén L, Hammarsjö
A, White I, Schoumans J, Läckgren G, Kockum CC and Nordenskjöld A:
22q11.2 microduplication in two patients with bladder exstrophy and
hearing impairment. Eur J Med Genet. 53:61–65. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zarate YA, Kogan JM, Schorry EK, Smolarek
TA and Hopkin RJ: A new case of de novo 11q duplication in a
patient with normal development and intelligence and review of the
literature. Am J Med Genet A. 143A:265–270. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Choi J, Lee H and Lee CG: Partial trisomy
of 11q23.3-q25 inherited from a maternal low-level mosaic
unbalanced translocation. Am J Med Genet A. 167A:1859–1864. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Göhring I, Blümlein HM, Hoyer J, Ekici AB,
Trautmann U and Rauch A: 6.7 Mb interstitial duplication in
chromosome band 11q24.2q25 associated with infertility, minor
dysmorphic features and normal psychomotor development. Eur J Med
Genet. 51:666–671. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Burnside RD, Lose EJ, Domínguez MG,
Sánchez-Corona J, Rivera H, Carroll AJ and Mikhail FM: Molecular
cytogenetic characterization of two cases with constitutional
distal 11q duplication/triplication. Am J Med Genet A.
149A:1516–1522. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kayhan G, Cavdarli B, Karaoguz Yirmibes M,
Percin EF, Oztürk Kaymak A, Biri A and Ergun MA: Molecular
karyotyping of an isolated partial trisomy 11q patient with
additional findings. Gene. 524:355–360. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ben-Abdallah-Bouhjar I, Mougou-Zerelli S,
Hannachi H, Ben-Khelifa H, Soyah N, Labalme A, Sanlaville D,
Elghezal H and Saad A: Phenotype and micro-array characterization
of duplication 11q22.1-q25 and review of the literature. Gene.
519:135–141. 2013. View Article : Google Scholar : PubMed/NCBI
|