Introduction
Spinal cord injury (SCI) involves a complex sequence
of pathological processes consisting of original injury, secondary
lesion and spinal restoration (1).
SCI is associated with severe neurological outcomes and limited
therapeutic options are available. The pathogenesis of SCI involves
altered blood circulation, abnormal distribution of bioactive
substances and production of free radicals (2). Anoxemia following the development of
SCI leads to tissue destruction, and results in a variety of
vascular pathologies resulting in acute arterial occlusion,
surgical interventions requiring clamping, trauma-causing ischemia,
transplantation, and shock (2–4).
Microglial cells are resident immune cells of the
central nervous system (CNS) that continuously survey the
environment and respond to alterations in cellular homeostasis
resulting from infection, hypoxia, stroke, and other stimuli. It
has previously been demonstrated that activated microglial cells
initiate neuronal and oligodendrocyte injury and exacerbate a
variety of neurological conditions by secreting pro-inflammatory
molecules including nitric oxide, tumor necrosis factor-alpha
(TNF-α), and interleukin-1β (IL-1β) (5–7).
Inflammation is important in protecting the CNS from infection, and
directly or indirectly controls the sequelae of SCI. The release of
inflammatory cytokines and chemokines from spinal cord cells in or
near the lesion initiates the inflammatory response in the CNS
(8,9).
Hypoxia-inducible factor 1 (HIF-1) is an important
regulator under conditions of hypoxia (10) and is also important in controlling
the neurological outcomes following ischemic stroke due to its
ability to regulate a variety of genes (11). HIF-1α is degraded in cells under
normoxic conditions, however is significantly upregulated during
hypoxia. The accumulation of HIF-1α is important in stimulating
angiogenesis, glycolysis and erythropoiesis by upregulating the
expression of downstream target genes, which are involved in
cellular tolerance to hypoxia and tissue vascularity, thus helping
the injured tissue to heal (12).
However, various studies have revealed that hypoxia may not only
directly damage neurons but may promote neuronal injury indirectly
via activation of microglial cells via the regulation of HIF-1α
during cerebral ischemia (13,14).
Autophagy is an intracellular catabolic pathway,
which degrades cell components, toxic aggregates and damaged
organelles, and recycles them as basic building blocks for the
efficient maintenance of cellular homeostasis (15). Autophagy is important for the
elimination of pathogens and production of cytokines from
macrophages (16,17). Autophagy controls inflammation
through interactions with immune signaling pathways and regulates
the secretion of molecular mediators of inflammation (18,19).
An appropriate level of autophagy contributes to neuroprotection,
whereas inappropriate levels may induce cell death (20,21).
Therefore, it was hypothesized that HIF-1α
regulation of autophagy is important in microglial cells during
SCI. However, the formation and regulation of autophagy in
microglial cells, and its effect on the production of
pro-inflammatory and cytotoxic factors under conditions of hypoxia
remain to be elucidated. The present study investigated the
relationship and mechanism of action of HIF-1α and autophagy in
microglial cells during the development of SCI.
Materials and methods
Reagents and antibodies
High glucose Dulbecco's modified Eagle's medium
(DMEM/H-G) was purchased from Hyclone Laboratories; GE Healthcare
Life Sciences (Logan, UT, USA). Fetal bovine serum (FBS) was
purchased from Gibco; Thermo Fisher Scientific, Inc. (Waltham, MA,
USA). The 4′,6-diamidino-2-phenylindole (DAPI) 0.25% trypsin-EDTA,
Cell Counting Kit (CCK-8), protein extraction kit including
radioimmunoprecipitation assay buffer (RIPA), and bicinchoninic
acid (BCA) protein assay kit were purchased from Beyotime Institute
of Biotechnology (Haimen, China). Dimethyl sulfoxide (DMSO) and
bovine serum albumin (BSA) were purchased from Biosharp
Biotechnology Co. (Hefei, China). 2-Methoxyestradiol (2ME2) was
purchased from Selleck Chemicals (Houston, TX, USA). Antibodies
against HIF-1α (cat. no. ab179483), microtubule-associated protein
1A/1B-light chain 3 (LC-3) phosphatidylethanolamine conjugate
(LC3-II) (cat. no. ab51520), and Beclin-1 (cat. no. ab207612) were
obtained from Abcam (Cambridge, UK). Antibodies against β-actin
(cat. no. GB13001-3), and tetramethylrhodamine (TRITC)-conjugated
secondary antibody (cat. no. 111-095-003) were obtained from
Sevicebio Technology (Wuhan, China).
Isolation of spinal microglial
cells
Spinal microglial cells were isolated by Percoll
density gradient centrifugation as previously described (22). A total of 20 intact male 1-day old
mice were provided by the animal experimental center of The Second
Military Medical University (Shanghai, China). Uniform commercial
diets were purchased from the animal experimental center of The
Second Military Medical University (Shanghai, China). Mice were
housed individually with feed and tap water available ad libitum at
25°C and 70% humidity under a 12/12 h light/dark cycle and were
sacrificed by CO2 asphyxia. Mice were placed into a box
in which euthanasia took place, the box was made of glass and had a
volume of 18 l (23×26×33 cm). A rising concentration of
CO2 was released into the cage by a tube at a rate of
3.6 l/min, the final concentration of CO2 was 100%. The
time taken to observe loss of consciousness and respiratory arrest
of mice was at least 5 min both in the cage and out of the cage.
Lumbar enlargements of the spinal cords of 1-day old mice were
digested with 0.1% trypsin. The study was approved by the ethics
committee of the Second Military Medical University (Shanghai,
China; no. 13071002124). Spinal cords were ground briefly in 4 ml
ice-cold Hanks, balanced salt solution (Beyotime Institute of
Biotechnology, Haimen, China) and filtered through a nylon mesh
(200 mesh; 74 µm) to remove astrocytes and undigested tissue. The
cells were seeded into flasks coated with poly-L-lysine and
cultured with DMEM supplemented with 10% FBS at 37°C with 5%
CO2. Culture media were replenished twice per week for 2
weeks. Microglial cells were detached by gentle shaking on a
horizontal shaker at 120 rpm for 3 h at 37°C, centrifuged at 800 ×
g for 10 min at 20°C, then resuspended in fresh DMEM supplemented
with 10% FBS and plated at a final density of 5×105
cells/ml in a six-well culture plate at 37°C with 5%
CO2. The cell purity was determined by
immunohistochemical (IHC) staining using microglial cell specific
antibody CD11b (cat. no. ab75476; rabbit against rat and mouse;
1:100; Abcam). The microglial cell cultures used were >95%
pure.
BV2 cell culture and hypoxic
conditions of microglial cells
Due to the limited number of primary culture cells
available, BV2 cells were used as substitute for the studies of
hypoxia-induced mechanisms in microglial cells, where stated. BV2
cells were obtained from the Department of Neurobiology, Second
Military Medical University (Shanghai, China). BV2 cells were
cultured in DMEM/H-G supplemented with 10% FBS, 50 U/l penicillin
and 50 mg/ml streptomycin at 37°C in a humidified atmosphere
incubator containing 5% CO2. BV2 is an immortalized
microglial cell line that has been reported to share numerous
characteristics exhibited by primary microglial cells (23,24).
For exposure to hypoxic conditions, the culture
medium was replaced, followed by appropriate treatment prior to
cell exposure to hypoxia in a chamber, containing a 1%
O2/5% CO2/94% N2 gas mixture for
the indicated time period. BV2 cells were pretreated with 3µM 2ME2
or transfected with 50 nM HIF-1α siRNA prior to exposure to
hypoxia.
Inhibitor and small interfering
(si)RNA treatment
Cells were exposed to the HIF-1α inhibitor 2ME2 to
determine the mechanisms of hypoxia-induced autophagy. BV2 cells
were seeded in six-well plates and pretreated with 3 µM 2ME2 for 6
h prior to hypoxia exposure. These pretreated BV2 cells were
exposed to hypoxia for 3, 6, 9, 12 and 24 h. Supernatants from cell
cultures were collected and assayed for secreted cytokines, and
cell pellets were used for RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis. For transfection with siRNA, cells were plated in
six-well plates at 5×105 cells/well. Plated cells were
grown in DMEM supplemented with 10% FBS overnight, and transfected
with 50 nM HIF-1α siRNA (5′-CTACTCAGGACACAGATTTAGACTTGGAG-3) or
negative control siRNA (5′-CAGCAACCAGGTGACCGTG-3′) obtained from
Guangzhou Ribobio Co. Ltd., Guangzhou, China, using the
transfection reagent Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific Inc.) according to the manufacturer's protocol, 24 h
prior to treatment with drugs.
CCK8 assay
CCK8 assays were performed in 96-well plates (100 µl
cells, 5×106/ml suspension/well) according to the
manufacturer's protocol. The cells were incubated with 10 µl CCK
solution following the indicated treatment by exposing them to
hypoxia in a chamber filled with a gas mixture of 1%
O2/5% CO2/94% N2. Absorbance was
measured at a wavelength of 450 nm using a microplate reader
(Bio-Rad laboratories Inc., Hercules, CA, USA) at 1–4 h. Cell
viability was expressed as a percentage of the value of the control
cultures, following subtraction of the background absorbance.
Evaluation of cell death by flow
cytometry
Cells were pretreated with 3 µM 2ME2 for 6 h or
transfected with 50 nM HIF-1α siRNA prior to hypoxia exposure for
3, 6, 9, 12 and 24 h. Following exposure to hypoxia and drugs,
cells were washed twice with phosphate buffered saline (PBS),
adjusted to a concentration of 1×106 cells/ml, fixed
with 70% ethanol on ice for 30 min, resuspended in 1 ml binding
buffer (Seebio Shanghai, China) and incubated at 4°C for 15 min in
the dark with 20 µg/ml annexin V-FITC and 50 µg/ml propidium
iodide. Samples were analyzed using Cytomics™ FC500
Summit Software, Version 4.3 (Beckman Coulter, Inc., Brea, CA, USA)
by flow cytometry using a BD FACSCanto II cytometer (BD
Biosciences, Franklin Lakes NJ, USA).
Transmission electron microscopy
(TEM)
Thin sections were cut with a diamond knife on an
Ultra 45°C (Daitome AG, Nidau, Switzerland) and examined using a
Tecnai G2 20 Twin transmission electron microscope (FEI, Hillsboro,
OR, USA). Uranium lead double staining (with 2% uranyl acetate
saturated aqueous solution, and then citrate for 15 min each).
Ultrastructural features of autophagy in microglial cells were
studied by TEM as previously described (25). Briefly, microglial cells were
harvested following exposure to hypoxia, washed with PBS, fixed
with 1% glutaraldehyde in 0.1 M cacodylate buffer for 2 h at 4°C,
then post-fixed with 1% OsO4 for 1.5 h in the same buffer at 20°C.
The samples were then washed, dehydrated, and embedded in 812
embedding medium (SPI Supplies, Inc, West Chester, PA, USA) at 20°C
for 12 h. Thin sections (60–80 nm) were cut with a diamond knife on
an Ultra 45°C (Daitome AG) and examined using a Tecnai G2 20 Twin
transmission electron microscope (FEI, Hillsboro, OR, USA).
IHC staining
BV2 cells were plated on cover slips in 24-well
plates and exposed to hypoxia or treated with drugs. IHC staining
of LC3-II was performed to determine expression level alterations
of LC3-II following exposure to hypoxia. Briefly, the cells were
fixed with 4% paraformaldehyde, permeabilized with 0.3% Triton
X-100, blocked with 5% goat serum for 40 min, and incubated with
rabbit anti-LC3-II antibodies (1:100; cat. no. ab51520) diluted in
PBS overnight at 4°C. Subsequently, cells were stained with the
tetramethylrhodamine-conjugated anti-rabbit IgG secondary antibody
(1:100; cat. no. 111-095-003) for 2 h at room temperature. The
cells were counterstained with DAPI for 5 min following rinsing
with PBS. The images were captured on a fluorescence microscope
(Olympus Corporation, Tokyo, Japan).
RT-qPCR
RT-qPCR was performed to measure mRNA expression
levels of HIF-1α, in BV2 cells. Total RNA was extracted using
TRIzol reagent (Thermo Fisher Scientific, Inc.), and the
reverse-transcription reactions were performed using a TaqMan
MicroRNA Reverse Transcription kit (Takara Bio, Inc., Otsu, Japan).
qPCR was performed using a standard SYBR Green PCR kit (Takara Bio,
Inc.) in a 7900 Real-time PCR system (Applied Biosystems, Thermo
Fisher Scientific Inc.) according to the manufacturer's protocol.
The thermocycling conditions used for the PCR were as follows: 95°C
for 30 sec, followed by 40 cycles of 95°C for 3 sec and 60°C for 30
sec. The sequences of primers used were as follows: HIF-1α, forward
5′-TGCTTGGTGCTGATTTGTGA-3′ and reverse 5′-GGTCAGATGATCAGAGTCCA-3′;
and β-actin, forward 5′-TTCCTTCCTGGGCATGGAGTCC-3′ and reverse
5′-TGGCGTACAGGTCTTTGCGG-3′. β-actin was used as a reference for
mRNAs and each sample was analyzed in triplicate. Relative levels
of gene expression were quantified using the 2−ΔΔCq
method (26).
Western blotting
Protein expression of Beclin-1 and LC3-II in treated
and untreated BV2 cells was analyzed by western blotting. Briefly,
BV2 cells were harvested at the indicated time, washed three times
with ice-cold PBS, and RIPA buffer was used to extract total and
nuclear proteins. The BCA protein assay was then used to determine
protein concentration. Following this, equal amounts of protein (20
µl 500 ng/ml) were separated on 10% SDS-PAGE minigels and
transferred to nitrocellulose membranes. The membranes were then
blocked using 5% skimmed milk for 60 min, and incubated with rabbit
primary antibodies against HIF-1α, Beclin-1 and LC3-II at a 1:200
dilution overnight at 4°C. Subsequently, the membranes were washed
three times for 10 min each with Tris-buffered saline with Tween-20
(0.05%; TBST) and incubated with the corresponding secondary
antibodies for 2 h at room temperature. Blots were washed three
times for 10 min each in TBST and scanned with the Bio-Rad western
blot system (Bio-Rad Laboratories, Inc.). Blots were washed three
times for 10 min each in TBST. Immunoblots were detected using an
enhanced chemiluminescence kit (cat. no. NC32109; Thermo Fisher
Scientific, Inc.). Autoradiographs were quantitated by the Bio-Rad
western blot system (Bio-Rad Laboratories, Inc.). Background
intensity was subtracted from each sample and normalized to the
β-actin loading control. Protein expression was quantified using an
Image Lab 4.0 software system (Bio-Rad Laboratories, Inc.).
Background intensity was subtracted from each sample and normalized
to the β-actin loading control.
ELISA
Supernatants were collected from cell cultures at
different time points following exposure to various stimuli. Cells
were stimulated with transfection of 50 nM HIF-1α siRNA or
pretreatment with 3 µM 2ME2 for 6 h prior to hypoxia exposure.
Secretion of cytokines TNF-α and IL-1β in the supernatants was
measured using ELISA kits (TNF-α, cat. no. 88-7340-22; IL-1β, cat.
no. 88-7010-77) according to the manufacturer's protocols
(eBioscience, Inc., San Diego, CA, USA). Absorbance was measured at
a wavelength of 405 nm on a Bio-Rad iMark microplate reader
(Bio-Rad Laboratories, Inc.). The protein concentrations (pg/ml)
were calculated from a standard curve.
Statistical analysis
Statistical analyses were conducted using SPSS
software, version 18.0 (SPSS, Inc., Chicago, IL, USA). All data are
expressed as the mean ± standard error of the mean. Differences
between groups were compared by one-way analysis of variance
followed by Bonferroni post-hoc multiple comparison test. Each
experiment included at least 3 replicates per condition. P<0.05
was considered to indicate a statistically significant
difference.
Results
Hypoxia-induces cell death and
inflammation in microglial cells
To elucidate the role of hypoxia in microglial cell
death, the primary microglial cells and BV2 cells were exposed to
hypoxia for 3, 6, 9, 12 and 24 h. Cell viability was measured by
CCK-8 assay, which is considered an effective method for the
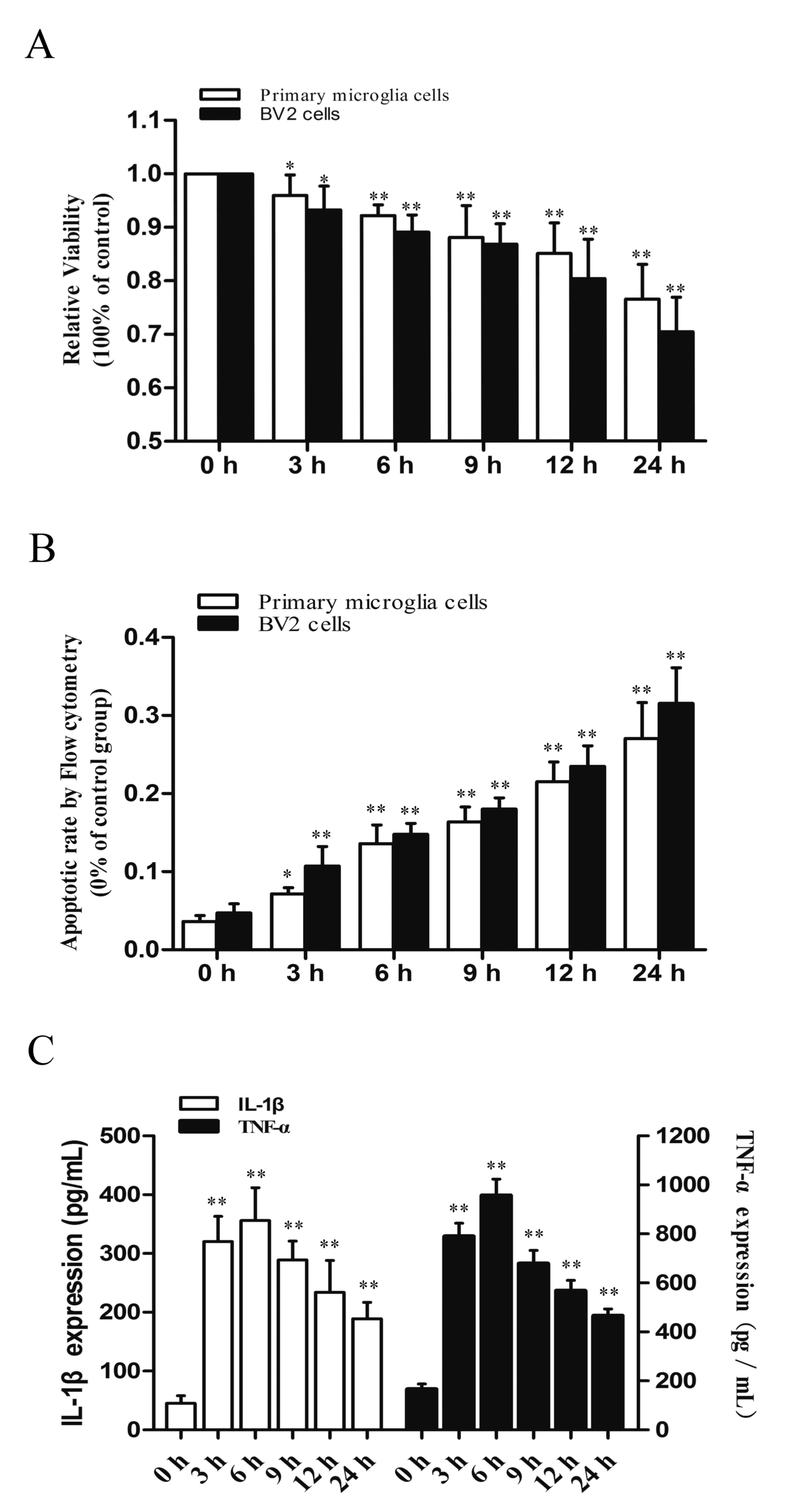
detection of cell death. As presented in Fig. 1A, statistically significant
time-dependent decreases in relative cell viability were observed
under hypoxic conditions in both cell types (P<0.001 vs. 0 h).
Similar results were obtained by flow cytometry: Hypoxia induced
apoptosis in microglial cells (P<0.001 vs. 0 h Fig. 1B). It has previously been
demonstrated that hypoxia rapidly induces cell death through
inflammation (27). To examine the
effects of hypoxia on neuroinflammation in microglial cells, the
expression of inflammatory cytokines IL-1β and TNF-α in
hypoxia-exposed microglial cells was investigated by ELISA. As
presented in Fig. 1C, hypoxia
significantly increased the protein levels of IL-1β and TNF-α,
reaching a peak at 6 h exposure compared with 0 h exposure
(P<0.001). Overall, the results demonstrated that hypoxia
induced cell death and inflammation in microglial cells.
Hypoxia-induces expression of HIF-1α
in BV2 cells
HIF-1α is expressed at a significantly high level
under hypoxic conditions and heterodimerizes with HIF-1β to form
HIF-1, following translocation into the nucleus (28). The present study investigated
whether hypoxia-induced cell death was HIF-1α-dependent. It was
observed that hypoxia significantly increased HIF-1α mRNA
expression levels at 3, 6, 9, 12 and 24 h compared with 0 h
(P<0.05, P<0.001, P<0.001, P<0.001 and P<0.001,
respectively; Fig. 2A). The
greatest level of expression of HIF-1α mRNA was observed at 6 h
compared with 0 h (P<0.001; Fig.
2A) and comparable results were observed by western blotting,
with hypoxia significantly increasing HIF-1α protein expression
levels at 3, 6, 9, 12 and 24 h compared with 0 h (P<0.001,
P<0.001, P<0.001, P<0.001 and P<0.05, respectively;
Fig. 2B). These data indicated
that hypoxia induced the expression of HIF-1α in microglial cells.
This effect was observed in groups exposed to hypoxia for 6 h,
which suggested that HIF-1α is important in hypoxia-induced cell
death.
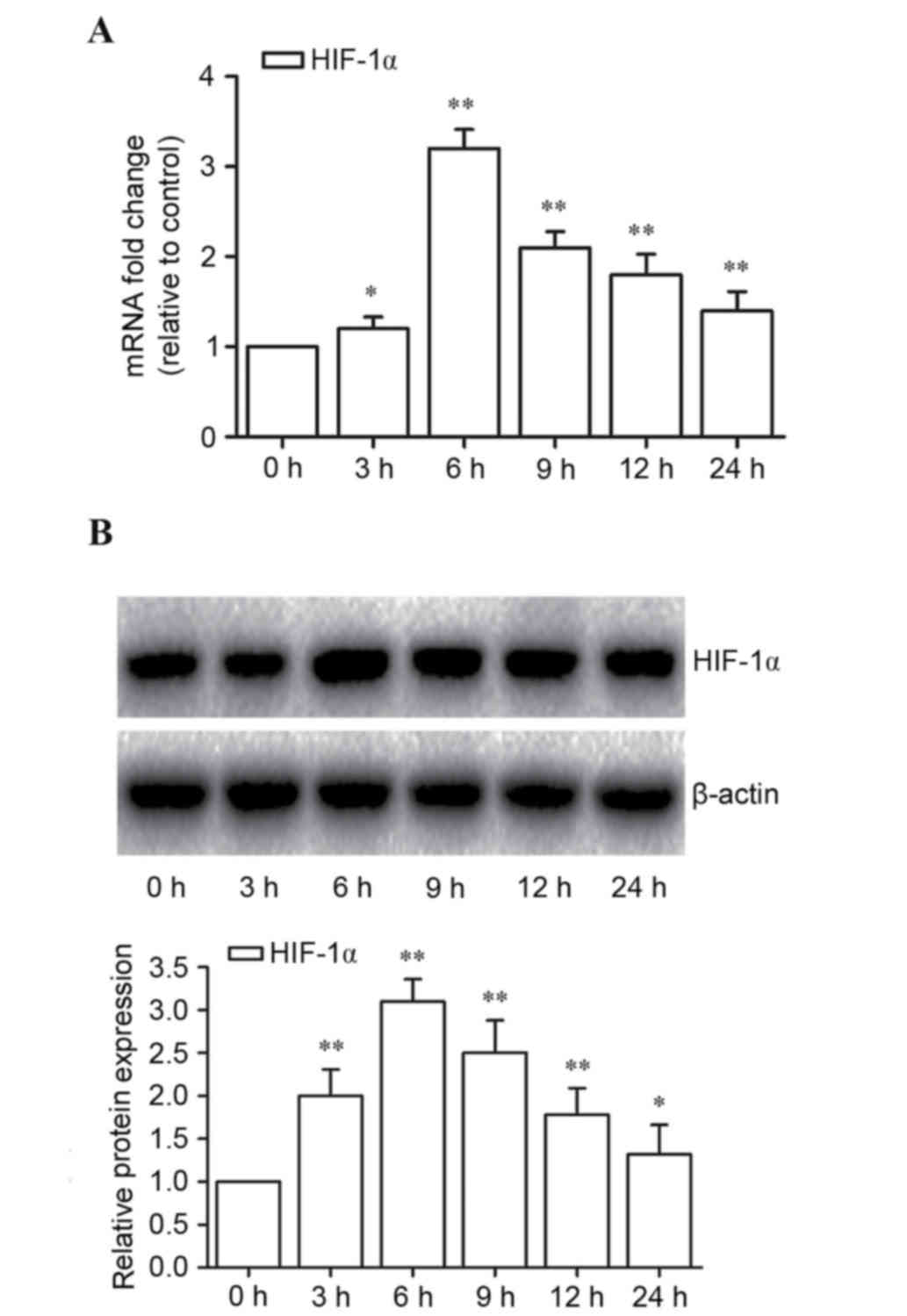 | Figure 2.HIF-1α mediates hypoxia-induced cell
death. (A) Reverse transcription-quantitative polymerase chain
reaction was used to determine mRNA expression levels of HIF-1α in
microglia cells following exposure to hypoxic conditions for 0, 3,
6, 9, 12 and 24 h. (B) HIF-1α protein expression levels in
microglia were detected by western blot assay following exposure to
hypoxic conditions for 0, 3, 6, 9, 12 and 24 h, and quantified
relative to β-actin. Data are presented as the mean ± standard
deviation of three independent experiments. *P<0.05 and
**P<0.001 vs. 0 h. HIF-1α, hypoxia-inducible factor 1-α. |
HIF-1α mediates microglial cell
autophagy induced by hypoxia in BV2 cells
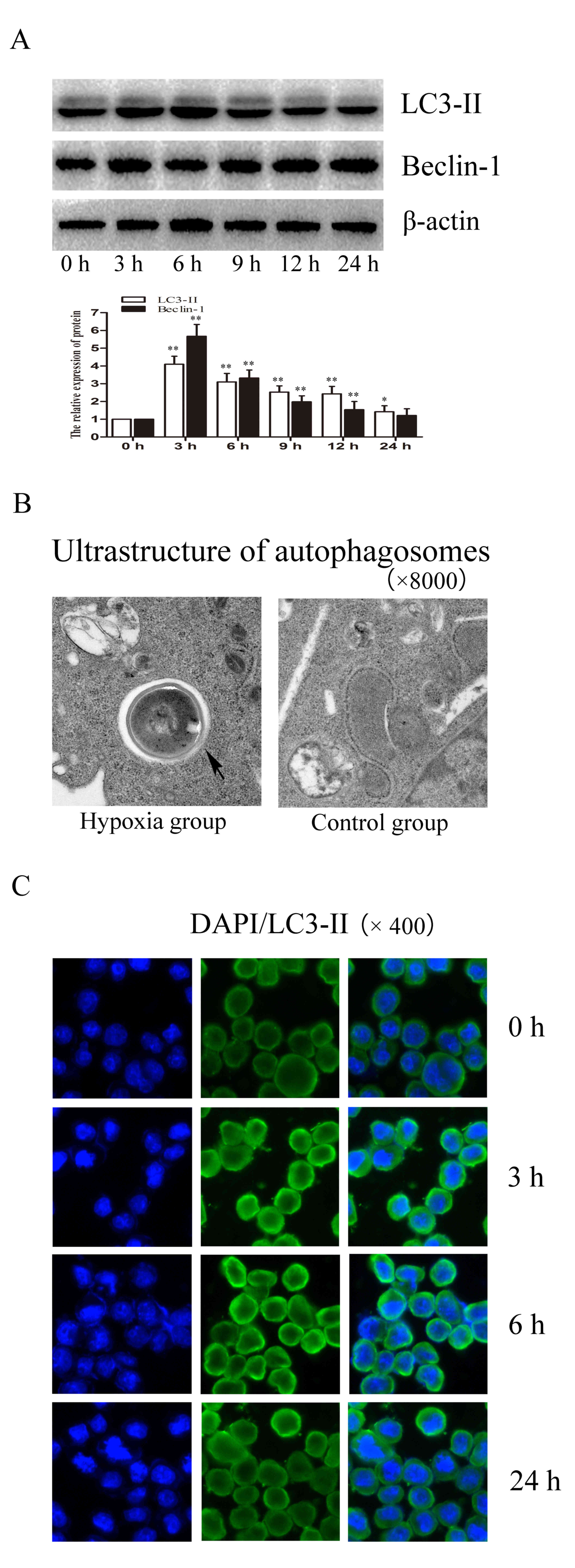
It has previously been suggested that SCI-associated
hypoxia may induce autophagy in microglial cells (29,30).
Beclin-1 and LC-3 are characteristic marker proteins of autophagy.
The expression levels of LC3-II and Beclin-1 were investigated to
determine whether autophagy is induced as a result of the
expression of HIF-1α following exposure to hypoxia. As presented in
Fig. 3A, the protein expression
levels of LC3-II and Beclin-1 reached their peak in hypoxic cells
after 3 h hypoxia compared with 0 h (P<0.001). Ultrastructural
alterations in hypoxia-treated microglial cells were examined and
compared with controls without hypoxia treatment (Fig. 3B). Closed arrows indicate the
presence of autophagosomes in hypoxia-treated microglial cells
(Fig. 3B). This indicates high
expression of autophagosomes in hypoxia-treated microglial cells.
As presented in Fig. 3C, the
protein accumulation of LC3-II visualized by immunofluorescence was
visibly increased in hypoxic cells compared with 0 h hypoxia
control.
HIF-1α mediates hypoxia-induced cell
death and inflammation via autophagy in BV2 cells
The previous results illustrate that hypoxia induces
a complete autophagic response in microglial cells, and HIF-1α may
mediate microglial cell autophagy under conditions of hypoxia.
Microglial cells were exposed to the HIF-1α inhibitor 2ME2 or
silenced with HIF-1α siRNA (100 nM each) for 6 h, to investigate
whether hypoxia-induced autophagy promoted microglial cell survival
or death, and to explore the associated mechanism of action. The
mRNA and protein expression levels of the HIF-1α target were
evaluated by RT-qPCR and western blotting assays respectively. Cell
death and inflammation induced by hypoxic conditions in cells with
inhibited HIF-1α were additionally evaluated.
At each time point, HIF-1α inhibition and HIF-1α
silencing significantly decreased hypoxia-induced HIF-1α mRNA
expression compared with the uninhibited control at the same time
point (2ME2 3 and 12 h, P<0.05 and P<0.05, respectively;
HIF-1α siRNA 3 and 12 h, P<0.05 and P<0.05, respectively;
Fig. 4A). In addition, at each
time point, HIF-1α inhibition and HIF-1α silencing visibly
decreased hypoxia-induced HIF-1α protein expression compared with
the uninhibited control at the same time point (Fig. 4B). LC3-II protein expression levels
were significantly reduced at 3 and 12 h in BV2 cells following
transfection with siRNAs targeting HIF-1α, compared with controls
at the same time points (P<0.05 and P<0.05, respectively,
Fig. 4C) and Beclin-1 protein
expression levels were significantly reduced at 3 h compared with
controls at 3 h (P<0.05; Fig.
4C).
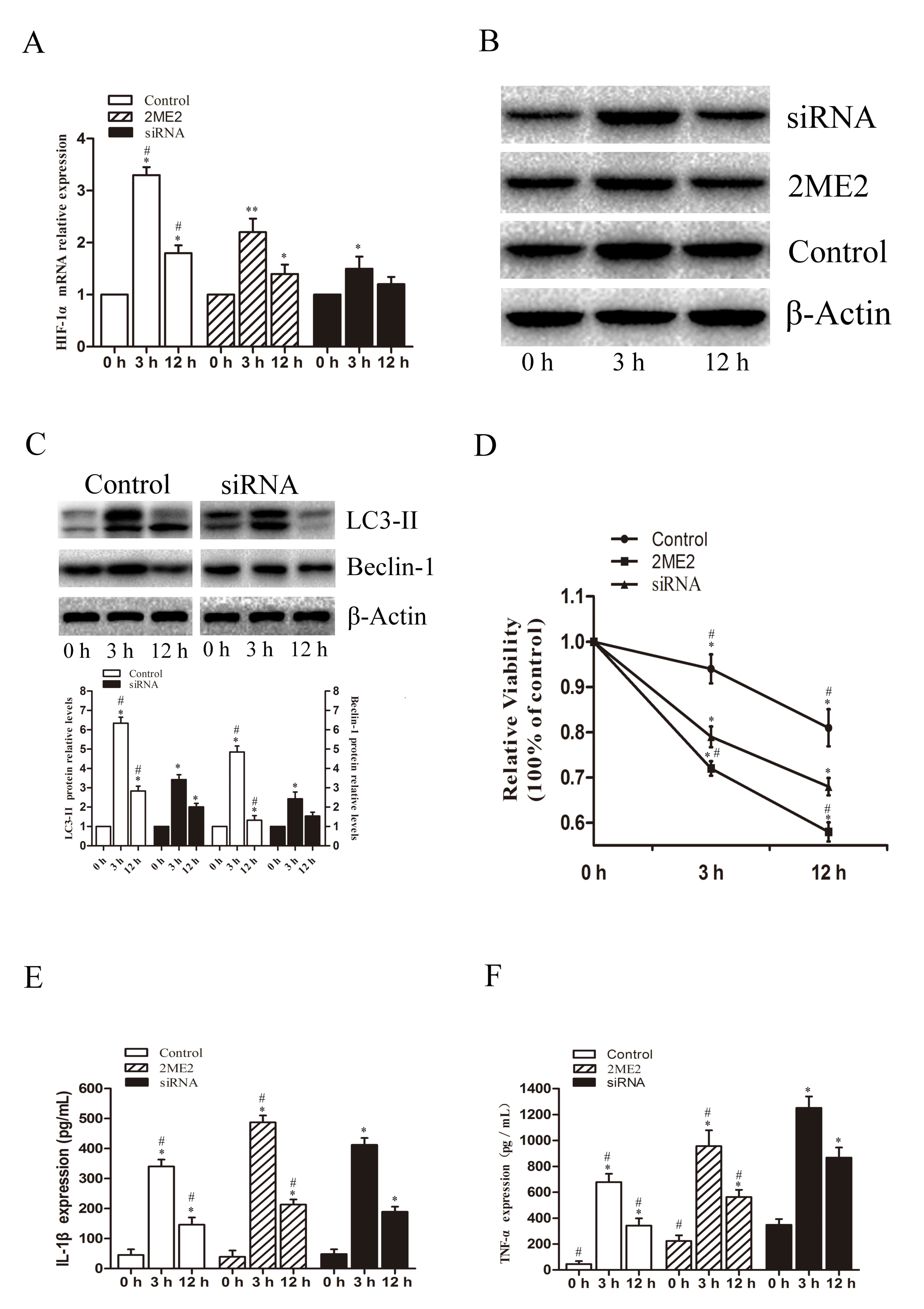 | Figure 4.HIF-1α mediates hypoxia-induced cell
death and inflammation by autophagy in BV2 cells. Detection of
efficiency of HIF-1α inhibitors and siRNAs on HIF-1α expression
following hypoxia in BV2 cells. Microglia cells were pretreated
with 3 µM 2ME2 or with siRNA targeting HIF-1α (100 nM) for 6 h, and
(A) mRNA and (B) protein expression levels of the target were
evaluated by quantitative polymerase chain reaction and western
blot assays, respectively. (C) Detection of the effect of HIF-1α
inhibition on LC3-II and Beclin-1 protein expression in BV2 cells
following hypoxia. Protein expression levels were evaluated by
western blot and quantified relative to β-actin. (D) Viability of
microglia cells exposed to the inhibitors following 0, 3 and 12 h
hypoxia was assessed by Cell Counting Kit-8 assay. Protein
expression levels of (E) IL-1β and (F) TNF-α were determined by
ELISA in microglia cells exposed to the inhibitors and 0, 3 and 12
h hypoxia. Data are expressed as the mean ± standard deviation of
three independent experiments. *P<0.05, **P<0.001 vs. the 0 h
control time point); #P<0.05, ##P<0.001
vs. the equivalent time point siRNA) HIF-1α, hypoxia-inducible
factor 1-α; LC3-II, microtubule-associated protein 1A/1B-light
chain 3 phosphatidylethanolamine conjugate; IL-1β, interleukin-β;
TNF-α, tumor necrosis factor-α; siRNA, small interfering RNA; 2ME2,
2-methoxyestradiol. |
Additional studies revealed that chemical inhibition
of HIF-1α or HIF-1α siRNA knockdown decreased hypoxia-induced cell
viability at 3 and 12 h compared with controls at the same time
points (2ME2 3 and 12 h, P<0.05 and P<0.05, respectively;
HIF-1α siRNA 3 and 12 h, P<0.05 and P<0.05, respectively;
Fig. 4D). This effect was observed
to be associated with alterations in the expression of inflammatory
cytokines in BV2 cells: the production of IL-1β was significantly
increased in BV2 cells treated with the inhibitor 2ME2 and
transfected with siRNAs targeting HIF-1α at 3 and 12 h compared
with controls without pre-treatment at the same time points (2ME2 3
and 12 h, P<0.05 and P<0.05, respectively; HIF-1α siRNA 3 and
12 h, P<0.05 and P<0.05, respectively; Fig. 4E) and TNF-α expression was also
significantly increased in BV2 cells treated with the inhibitor
2ME2 and transfected with siRNAs targeting HIF-1α at 3 and 12 h
compared with controls without pre-treatment at the same time
points (2ME2 3 and 12 h, P<0.05 and P<0.05, respectively;
HIF-1α siRNA 3 and 12 h, P<0.05 and P<0.05, respectively;
Fig. 4F).
These data suggested that inhibition of HIF-1α
decreased cell death via controlled overexpression of inflammatory
cytokines in BV2 cells. HIF-1α may therefore mediate the autophagic
death of microglial cells via regulation of autophagy-associated
gene expression and cytokine release.
Discussion
In the present study, the effects and mechanism of
action of hypoxia-induced autophagic cell death in microglial cells
were investigated. The results demonstrated that hypoxia induced
cell death and inflammation in microglial cells. HIF-1α mediated
hypoxia tolerance in hypoxia-induced autophagic cell death in
microglial cells. The present study suggested that effects may be
associated with modulating the expression of autophagy-associated
genes and the inflammatory response following hypoxia in microglial
cells.
SCI, which is characterized by primary physical and
secondary damage, leads to severe consequences including paralysis,
intense pain and progressive neurological damage (31). Primary spinal cord injuries
attributed to mechanical forces are characterized by acute
hemorrhage and ischemia, whereas secondary injuries are associated
with an inflammatory response, further characterized by further
destruction of neuronal and glial cells, resulting in the release
of reactive oxygen species and pro-inflammatory cytokines (32,33).
Macrophages in the CNS derived from blood monocytes or resident
microglial cells, and microglial cells, are important in
maintaining and restoring CNS homeostasis. Macrophages accumulate
within the epicenter of the injury and the hematoma of the injured
spinal cord under pathological conditions, and may be important in
the progression of inflammation (34). Pro-inflammatory cytokines including
TNF-α, IL-1β, and IL-6 and inflammatory mediators, including
inducible nitric oxide synthase and cyclooxygenase-2 are rapidly
upregulated following SCI (35,36).
However, the mechanisms by which microglial cells control
pro-inflammatory cytokines during SCI have not been fully
elucidated.
To investigate the effect of hypoxia on cell
viability and pro-inflammatory cytokines in microglial cells, the
present study first performed a cell death assay and detected the
secretion of pro-inflammatory cytokines. These data indicated that
hypoxia gradually induced cell death along with an increase in
inflammation, and this was consistent with data from previous
reports (37,38).
HIF-1 is a heterodimeric transcription factor that
contains the protein HIF-1α and an arylhydrocarbon receptor nuclear
translocator (HIF-1β) (39). The
availability of HIF-1 is primarily determined by levels of HIF-1α,
which is stably expressed. The expression and activity of the
HIF-1α subunit are regulated by the cellular oxygen concentration.
HIF-1α is expressed at a high level under hypoxic conditions, and
heterodimerizes with HIF-1β to form HIF-1 following translocation
to the nucleus. At an early stage of hypoxia, induction of HIF-1
may protect cells from destruction (40,41).
The present study investigated whether HIF-1α was
involved in cell death induced by hypoxia. The expression of HIF-1α
increased in a time-dependent manner under hypoxic conditions.
During cerebral anoxemia, the activation of HIF-1α is an important
activator of microglial cells (42). In addition, microglial cells were
exposed to a HIF-1α inhibitor and HIF-1α siRNA to evaluate the
association between hypoxic conditions and subsequent cell
death.
Autophagy is a cardinal cellular mechanism that
involves degradation and digestion of intracellular constituents by
lysosomes (43,44). Autophagy is important under
physiological conditions for numerous immune cells including
macrophages (16,45). It has previously been suggested
that autophagy has a cytoprotective function against cell death
(46) and may contribute to
cytoprotection in neurodegenerative disease and traumatic brain
injury (47,48). Conversely, abrogated autophagy may
induce cell death (49,50). Autophagy may lead to non-apoptotic
programmed cell death, known as autophagic cell death. LC-3, a
homolog of yeast autophagy-related protein 8 (Atg8), is essential
for autophagy: It is important in the formation of the
autophagosome and is considered to be another specific marker
protein to monitor autophagy (51). LC3-II is a widely used marker for
autophagy; during autophagy, the cytosolic LC3-I form is converted
to an autophagosome-bound LC3-II form (52,53).
Beclin-1 is another autophagy-associated protein, which is
important in regulating the development of precursors of
autophagosomes from other autophagy-associated proteins (54). Beclin-1 and LC-3 are, therefore,
characteristic markers of autophagy (55).
Therefore, to further determine whether autophagy is
induced under hypoxic conditions, the expression of LC3-II and
Beclin-1 was investigated. Hypoxia induced a complete autophagic
response in microglial cells. In addition, autophagy was observed
by inhibiting HIF-α activation in microglial cells via a HIF-1α
inhibitor or HIF-1α siRNA. It was observed that HIF-1α mediated
microglial cell autophagy under hypoxic conditions.
Neuroinflammation is characterized by the activation
of microglial cells, and contributes to the development of numerous
neurological conditions, including CNS infections, ischemic stroke,
neurodegenerative diseases and anesthetic neurotoxicity, through
the production of various pro-inflammatory mediators, which promote
neuronal cell death (38,56). Autophagy controls inflammation
through interactions with immune signaling pathways and regulates
secretion of inflammatory molecular mediators (19,57).
However, the pathways of microglial cells controlling
pro-inflammatory cytokines under hypoxic conditions during SCI have
not yet been elucidated. The present study examined the extent of
cell death and inflammation induced by hypoxia. The results
demonstrated that HIF-1α was important in hypoxia-induced cell
death and mediated microglial cell autophagic cell death via
regulation of autophagy-associated genes and cytokine release.
In conclusion, the present study demonstrated that
hypoxia enhanced the expression of HIF-1α in microglial cells and
resulted in autophagic cell death through the activation of
microglial cells. Expression of HIF-1α inhibited the overexpression
of the inflammatory cytokine in microglial cells at an early stage
of hypoxia. In addition, various autophagic factors including
LC3-II and Beclin-1 were regulated by HIF-1α in microglial cells,
which mediated the expression of a variety of inflammatory
cytokines. HIF-1α stimulated the production inflammatory cytokines
via autophagic factors in microglial cells under hypoxic
conditions, thus enhancing the tolerance of microglial cells to
hypoxia during hypoxic conditions, which may represent a potential
therapeutic approach against diseases, including ischemic/hypoxic
SCI.
References
|
1
|
Amar AP and Levy ML: Pathogenesis and
pharmacological strategies for mitigating secondary damage in acute
spinal cord injury. Neurosurgery. 44:1027–1040. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fleming JC, Norenberg MD, Ramsay DA,
Dekaban GA, Marcillo AE, Saenz AD, Pasquale-Styles M, Dietrich WD
and Weaver LC: The cellular inflammatory response in human spinal
cords after injury. Brain. 129:3249–3269. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dolan EJ and Tator CH: The effect of blood
transfusion, dopamine, and gamma hydroxybutyrate on posttraumatic
ischemia of the spinal cord. J Neurosurg. 56:350–358. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Webb AA, Ngan S and Fowler JD: Spinal cord
injury I: A synopsis of the basic science. Can Vet J. 51:485–492.
2010.PubMed/NCBI
|
|
5
|
Brown GC and Neher JJ: Inflammatory
neurodegeneration and mechanisms of microglial killing of neurons.
Mol Neurobiol. 41:242–247. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Magazine HI, Liu Y, Bilfinger TV,
Fricchione GL and Stefano GB: Morphine-induced conformational
changes in human monocytes, granulocytes, and endothelial cells and
in invertebrate immunocytes and microglia are mediated by nitric
oxide. J Immunol. 156:4845–4850. 1996.PubMed/NCBI
|
|
7
|
Perry VH, Nicoll JA and Holmes C:
Microglia in neurodegenerative disease. Nat Rev Neurol. 6:193–201.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Carlson SL, Parrish ME, Springer JE, Doty
K and Dossett L: Acute inflammatory response in spinal cord
following impact injury. Exp Neurol. 151:77–88. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Donnelly DJ and Popovich PG: Inflammation
and its role in neuroprotection, axonal regeneration and functional
recovery after spinal cord injury. Exp Neurol. 209:378–388. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
De Lemos ML, de la Torre AV, Petrov D,
Brox S, Folch J, Pallàs M, Lazarowski A, Beas-Zarate C, Auladell C
and Camins A: Evaluation of hypoxia inducible factor expression in
inflammatory and neurodegenerative brain models. Int J Biochem Cell
Biol. 45:1377–1388. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Semenza GL: Expression of
hypoxia-inducible factor 1: Mechanisms and consequences. Biochem
Pharmacol. 59:47–53. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yeo EJ, Chun YS and Park JW: New
anticancer strategies targeting HIF-1. Biochem Pharmacol.
68:1061–1069. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bergeron M, Gidday JM, Yu AY, Semenza GL,
Ferriero DM and Sharp FR: Role of hypoxia-inducible factor-1 in
hypoxia-induced ischemic tolerance in neonatal rat brain. Ann
Neurol. 48:285–296. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sheldon RA, Osredkar D, Lee CL, Jiang X,
Mu D and Ferriero DM: HIF-1 alpha-deficient mice have increased
brain injury after neonatal hypoxia-ischemia. Dev Neurosci.
31:452–458. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang Z and Klionsky DJ: Mammalian
autophagy: Core molecular machinery and signaling regulation. Curr
Opin Cell Biol. 22:124–131. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Singh SB, Davis AS, Taylor GA and Deretic
V: Human IRGM induces autophagy to eliminate intracellular
mycobacteria. Science. 313:1438–1441. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee J, Kim HR, Quinley C, Kim J,
Gonzalez-Navajas J, Xavier R and Raz E: Autophagy suppresses
interleukin-1β (IL-1β) signaling by activation of p62 degradation
via lysosomal and proteasomal pathways. J Biol Chem. 287:4033–4040.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oka T, Hikoso S, Yamaguchi O, Taneike M,
Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, et
al: Mitochondrial DNA that escapes from autophagy causes
inflammation and heart failure. Nature. 485:251–255. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Korkaya H and Wicha MS: Inflammation and
autophagy conspire to promote tumor growth. Cell Cycle.
10:2623–2624. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Puyal J and Clarke PG: Targeting autophagy
to prevent neonatal stroke damage. Autophagy. 5:1060–1061. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rosello A, Warnes G and Meier UC: Cell
death pathways and autophagy in the central nervous system and its
involvement in neurodegeneration, immunity and central nervous
system infection: To die or not to die-that is the question. Clin
Exp Immunol. 168:52–57. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Willemen HL, Eijkelkamp N, Wang H, Dantzer
R, GW II Dorn, Kelley KW, Heijnen CJ and Kavelaars A:
Microglial/macrophage GRK2 determines duration of peripheral
IL-1beta-induced hyperalgesia: Contribution of spinal cord CX3CR1,
p38 and IL-1 signaling. Pain. 150:550–560. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bocchini V, Mazzolla R, Barluzzi R, Blasi
E, Sick P and Kettenmann H: An immortalized cell line expresses
properties of activated microglial cells. J Neurosci Res.
31:616–621. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sheng W, Zong Y, Mohammad A, Ajit D, Cui
J, Han D, Hamilton JL, Simonyi A, Sun AY, Gu Z, et al:
Pro-inflammatory cytokines and lipopolysaccharide induce changes in
cell morphology, and upregulation of ERK1/2, iNOS and sPLA (2)-IIA
expression in astrocytes and microglia. J Neuroinflammation.
8:1212011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rabinovich GA, Riera CM and Iribarren P:
Granulocyte-macrophage colony-stimulating factor protects dendritic
cells from liposome-encapsulated dichloromethylene
diphosphonate-induced apoptosis through a Bcl-2-mediated pathway.
Eur J Immunol. 29:563–570. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
gene expression data using real-time quantitative PCR and the
2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ratcliffe PJ, O'Rourke JF, Maxwell PH and
Pugh CW: Oxygen sensing, hypoxia-inducible factor-1 and the
regulation of mammalian gene expression. J Exp Biol. 201:1153–1162.
1998.PubMed/NCBI
|
|
28
|
Michel G, Minet E, Ernest I, Durant F,
Remacle J and Michiels C: Molecular modeling of the
hypoxia-inducible factor 1 (HIF-1). Theoretical Chemistry Accounts.
101:51–56. 1999. View Article : Google Scholar
|
|
29
|
Walker CL, Walker MJ, Liu NK, Risberg EC,
Gao X, Chen J and Xu XM: Systemic bisperoxovanadium activates
Akt/mTOR, reduces autophagy, and enhances recovery following
cervical spinal cord injury. PloS One. 7:e300122012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kanno H, Ozawa H, Sekiguchi A and Itoi E:
The role of autophagy in spinal cord injury. Autophagy. 5:390–392.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Oyinbo CA: Secondary injury mechanisms in
traumatic spinal cord injury: A nugget of this multiply cascade.
Acta Neurobiol Exp (Wars). 71:281–299. 2011.PubMed/NCBI
|
|
32
|
Hayashi M, Ueyama T, Nemoto K, Tamaki T
and Senba E: Sequential mRNA expression for immediate early genes,
cytokines, and neurotrophins in spinal cord injury. J Neurotrauma.
17:203–218. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Beattie MS: Inflammation and apoptosis:
linked therapeutic targets in spinal cord injury. Trends Mol Med.
10:580–583. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang YK, Liu JT, Peng ZW, Fan H, Yao AH,
Cheng P, Liu L, Ju G and Kuang F: Different TLR4 expression and
microglia/macrophage activation induced by hemorrhage in the rat
spinal cord after compressive injury. J Neuroinflammation.
10:1122013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang CX, Nuttin B, Heremans H, Dom R and
Gybels J: Production of tumor necrosis factor in spinal cord
following traumatic injury in rats. J Neuroimmunol. 69:151–156.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yune TY, Chang MJ, Kim SJ, Lee YB, Shin
SW, Rhim H, Kim YC, Shin ML, Oh YJ, Han CT, et al: Increased
production of tumor necrosis factor-alpha induces apoptosis after
traumatic spinal cord injury in rats. J Neurotrauma. 20:207–219.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang L, Zhang J, Yang L, Dong Y, Zhang Y
and Xie Z: Isoflurane and sevoflurane increase interleukin-6 levels
through the nuclear factor-kappa B pathway in neuroglioma cells. Br
J Anaesth. 110 Suppl 1:i82–i91. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gonzalez H, Elgueta D, Montoya A and
Pacheco R: Neuroimmune regulation of microglial activity involved
in neuroinflammation and neurodegenerative diseases. J
Neuroimmunol. 274:1–13. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cao Y, Mao X, Sun C, Zheng P, Gao J, Wang
X, Min D, Sun H, Xie N and Cai J: Baicalin attenuates global
cerebral ischemia/reperfusion injury in gerbils via anti-oxidative
and anti-apoptotic pathways. Brain Res Bull. 85:396–402. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Graumann U, Reynolds R, Steck AJ and
Schaeren-Wiemers N: Molecular changes in normal appearing white
matter in multiple sclerosis are characteristic of neuroprotective
mechanisms against hypoxic insult. Brain Pathol. 13:554–573. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Schmid T, Zhou J and Brune B: HIF-1 and
p53: Communication of transcription factors under hypoxia. J Cell
Mol Med. 8:423–431. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kyotani Y, Ota H, Itaya-Hironaka A,
Yamauchi A, Sakuramoto-Tsuchida S, Zhao J, Ozawa K, Nagayama K, Ito
S, Takasawa S, et al: Intermittent hypoxia induces the
proliferation of rat vascular smooth muscle cell with the increases
in epidermal growth factor family and erbB2 receptor. Exp Cell Res.
319:3042–3050. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jiang T, Yu JT, Zhu XC, Wang HF, Tan MS,
Cao L, Zhang QQ, Gao L, Shi JQ, Zhang YD and Tan L: Acute metformin
preconditioning confers neuroprotection against focal cerebral
ischaemia by pre-activation of AMPK-dependent autophagy. Br J
Pharmacol. 171:3146–3157. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Urbanek T, Kuczmik W, Basta-Kaim A and
Gabryel B: Rapamycin induces of protective autophagy in vascular
endothelial cells exposed to oxygen-glucose deprivation. Brain Res.
1553:1–11. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nakahira K, Haspel JA, Rathinam VA, Lee
SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim
HP, et al: Autophagy proteins regulate innate immune responses by
inhibiting the release of mitochondrial DNA mediated by the NALP3
inflammasome. Nat Immunol. 12:222–230. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rubinsztein DC, DiFiglia M, Heintz N,
Nixon RA, Qin ZH, Ravikumar B, Stefanis L and Tolkovsky A:
Autophagy and its possible roles in nervous system diseases, damage
and repair. Autophagy. 1:11–22. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Erlich S, Alexandrovich A, Shohami E and
Pinkas-Kramarski R: Rapamycin is a neuroprotective treatment for
traumatic brain injury. Neurobiol Dis. 26:86–93. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sarkar S, Ravikumar B, Floto RA and
Rubinsztein DC: Rapamycin and mTOR-independent autophagy inducers
ameliorate toxicity of polyglutamine-expanded huntingtin and
related proteinopathies. Cell Death Differ. 16:46–56. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Clarke PG: Developmental cell death:
Morphological diversity and multiple mechanisms. Anat Embryol
(Berl). 181:195–213. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Larsen KE and Sulzer D: Autophagy in
neurons: A review. Histol Histopathol. 17:897–908. 2002.PubMed/NCBI
|
|
51
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kitanaka C and Kuchino Y:
Caspase-independent programmed cell death with necrotic morphology.
Cell Death Differ. 6:508–515. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Scarlatti F, Granata R, Meijer AJ and
Codogno P: Does autophagy have a license to kill mammalian cells?
Cell Death Differ. 16:12–20. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by Beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kanno H, Ozawa H, Sekiguchi A and Itoi E:
Spinal cord injury induces upregulation of Beclin 1 and promotes
autophagic cell death. Neurobiol Dis. 33:143–148. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Pais TF, Szegő ÉM, Marques O,
Miller-Fleming L, Antas P, Guerreiro P, De Oliveira RM, Kasapoglu B
and Outeiro TF: The NAD-dependent deacetylase sirtuin 2 is a
suppressor of microglial activation and brain inflammation. EMBO J.
32:2603–2616. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Green DR, Galluzzi L and Kroemer G:
Mitochondria and the autophagy-inflammation-cell death axis in
organismal aging. Science. 333:1109–1112. 2011. View Article : Google Scholar : PubMed/NCBI
|