Introduction
Hypoxia is a fundamental biological phenomenon that
is strongly associated with tissue damage and cell viability under
stress conditions. It is widely accepted that hypoxic foci are
present in the microenvironment during ischemic injuries, including
neurological (1), intestinal
(2), myocardial (3) and liver (4) damage. These affect mitochondrial
respiratory chain functions, mitochondrial enzymes and adenosine
triphosphate synthesis (5). In
addition, hypoxia develops in normal tissues following radiation
exposure and is associated with increased inflammatory corpuscle
accumulation and activation, oxidative stress, and profibrogenic
cytokine activity, thus contributing to radiation-induced normal
tissue injury (6,7). Reactive oxygen species
(ROS)-dependent apoptosis via attenuation of mitochondrial function
and signaling pathways, has been demonstrated to be a major cause
of hypoxia-associated tissue injury (8–11).
Insulin-like growth factor-1 (IGF-1) functions to
promote a survival and proliferation in specific tissues by
initiating signaling cascades following binding to extracellular
IGF-1 receptor (IGF-1R), which results in IGF-1 activation and
phosphorylation (12). IGF-1R is a
member of the tyrosine kinase receptor superfamily, which is
involved in the regulation of cell proliferation, differentiation,
and survival (12). A previous
study demonstrated that IGF-1R is involved in apoptosis induction
through the reduction of mitochondrial dysfunction (13). The protective mechanisms associated
with IGF-1R involve preservation of the mitochondrial membrane
potential and reduction of caspase-3 activity (13). A previous study indicated that
IGF-1R in the endothelium maintains the endothelial barrier by
stabilizing the vascular endothelial (VE)-protein tyrosine
phosphatase/VE-cadherin complex (14). Furthermore, decreased IGF-1R
expression impairs endothelial function and increases renal
fibrosis, which is associated with kidney disease (14). Similarly, IGF-1R is essential in
mediating IGF activity during neuronal cell development. IGF-1R in
neuronal cells is critically important for their survival following
hypoxic/ischemic (H/I) injury (1).
IGF-mediated upregulation of the neuronal cellular inhibitor of
apoptosis-1 and X-linked inhibitor of apoptosis protein, contribute
to IGF/IGF-1R protection against neuronal apoptosis following H/I
injury (1). In addition,
IGF/IGF-1R have been demonstrated to protect against intestinal and
cardiomyocyte ischemic-reperfusion (I/R) injury (2,3).
Hypoxia is one of the factors involved in the
regulation of the IGF system (15,16).
IGF-1R expression and concentration are altered when cells, such as
human hepatocytes and growth neuronal cones, are exposed to hypoxic
conditions (15,16). Additionally, IGF-1/IGF-1R protect
cultured human cells against a variety of injuries, such as
oxidative stress and hypoxia, through the activation of associated
proteins including nuclear factor-κB and cyclic adenosine
monophosphate-response element binding protein (17–19).
However, the mechanisms associated with its anti-apoptotic effects
remains unknown. The present study demonstrates an association
between IGF-1R and the phosphatidylinositol 3-kinase
(PI3K)/threonine protein kinase B (Akt)/mammalian target of
rapamycin mTOR signaling pathway and autophagy. In addition, these
results provide evidence supporting the protective role of IGF-1R
against oxidative stress under hypoxic conditions.
Materials and methods
Cell lines and reagents
R- and R+ cells were a gift from Dr. Yu Dong
(Soochow University, Suzhou, China). R- cells were fibroblast cell
lines derived from mouse embryos with targeted disruption of the
IGF-1R genes (20). R+ cells were
derived from R- cells following co-transfection with a human IGF-1R
expression plasmid and a pLHL4 plasmid carrying the hygromycin
resistance gene (20,21). All cells were cultured in
Dulbecco's modified Eagle's medium (Biowest, Nuaillé, France)
supplemented with 1% penicillin/streptomycin (P0781; Sigma-Aldrich;
Merck Millipore, Darmstadt, Germany) and 10% fetal bovine serum
(Biowest) in a free-gas exchange chamber with atmospheric air at
37°C. Hypoxia treatment (24 or 48 h) was performed in a tri-gas
incubator (YCP-50S; Huaxi Electronic Technology Co., Ltd., Hunan,
China) at 37°C, 5% CO2, 93% N2 and 2%
O2. The PI3K inhibitor LY294002 (Sigma-Aldrich; Merck
Millipore) was used for 24 h at 10 µM. The autophagy inhibitor
3-methyladenine (3MA; M9281-100MG; Sigma-Aldrich; Merck Millipore)
was used at a concentration of 5 µM for 24 h.
RNA isolation and quantitative
polymerase chain reaction (qPCR)
R- and R+ cells were grown to a number of
107 and then digested with trypsin. The total RNA was
isolated using a High Pure RNA Isolation kit (RNAprep Pure; Tiangen
Biotech Co., Ltd., Beijing, China) and to eliminate genomic DNA
contamination with with DNase I (Tiangen Biotech Co., Ltd.)
according to the manufacturer's instructions. Total RNA (500 ng)
was used as a template for reverse transcription reactions using
the PrimeScript RT Reagent kit (Perfect Real Time; Takara
Biotechnology Co., Ltd., Dalian, China), followed by qPCR analysis.
The PCR procedures were performed as follows: Predenaturing at 95°C
for 5 min followed by 40 cycles of amplifications by denaturing at
95°C for 15 sec, annealing for 30 sec at 64°C for IGF-1R and 60°C
for GAPDH, then extension at 72°C for 30 sec, followed by a final
extension step at 72°C for 10 min. Relative expression of target
genes was analyzed by the ∆∆Cq method (22). GAPDH was used as an internal
control. The following primers were used for quantitative PCR:
IGF-1R sense, 5′-TACAACTACGCCCTGGTCATC-3′, and antisense,
5′-CTTCTCACACATCGGCTTCTC-3′; GAPDH, sense,
5′-TGAAGGTCGGTGTGAACGGATTTGG-3′, and antisense,
5′-ACGACATACTCAGCACCGGCCTCAC-3′. IGF-1R expression was evaluated
using the LightCycler 480 SYBR Green I Master kit (Roche
Diagnostics, Basel, Switzerland).
Western blotting
Cells were grown to a number of 107 and were lysed
with radioimmunoprecipitation assay lysis buffer (Roche
Diagnostics) and total protein was quantified using the Pierce BCA
Protein assay kit (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) according to the manufacturer's instructions. Equal quantities
of protein (20 µg) were separated using SDS-PAGE on a 10% (w/v)
polyacrylamide gel, then were electrotransferred onto a BioTrace NC
Membrane (Pall Life Sciences, Port Washington, NY, USA). Blots on
NC membrane were blocked for 1 h with blocking buffer consisting of
5% (w/v) bovine serum albumin (Biowest) and 0.1% (v/v) Tween 20
(Sigma-Aldrich; Merck Millipore) in phosphate-buffered saline (PBS;
HyClone; GE Healthcare Life Sciences, Logan, UT, USA). The
antibodies used for western blotting were as follows: PI3K-110
(#4252S; 1:1,000; Cell Signaling Technology, Inc., Danvers, MA,
USA), phosphorylated (p)-Akt/Akt (#14293/#12178; 1:1,000; Cell
Signaling Technology, Inc.), p-mTOR/mTOR (#5536S/#2983S; 1:1,000;
Cell Signaling Technology, Inc.), and autophagy marker light chain
3 (M186-3; LC3; 1:1,000; Medical and Biological Laboratories Co.,
Ltd., Nagoya, Japan) and β-actin (#12620; 1:1,000; Cell Signaling
Technology, Inc.). Primary antibodies were incubated overnight at
4°C, followed by incubation with horseradish peroxidase-conjugated
secondary anti-rabbit (AP187R; 1:10,000; EMD Millipore, Billerica,
MA, USA) or anti-mouse IgG antibodies (GTX26709; 1:10,000; GeneTex,
Inc., Irvine, CA, USA) for 1 h at 37°C. Protein bands were detected
using the ECL Blotting Detection Reagent (Thermo Fisher Scientific,
Inc.), and images were quantified using the Chemioscope Mini system
(Bioshine, Shanghai, China).
Autophagy detection
Cells were grown to a number of 107, digested and
washed twice with PBS (pH 7.4; HyClone; Thermo Fisher Scientific,
Inc.), centrifuged at 175 × g for 5 min at 37°C, and resuspended in
a PBS. The autophagosomes were marked and stained using the Cyto-ID
Autophagy Detection kit (Enzo Biochem, Inc., New York, NY, USA) and
photographed using a fluorescence microscope (Nikon Corporation,
Tokyo, Japan). Fluorescence intensity was detected by flow
cytometry (FC 500 MPL; Beckman Coulter, Inc., Brea, CA, USA).
Apoptosis detection
Cells were grown to a number of 107, digested with
trypsin and washed twice with PBS (pH 7.4; HyClone; GE Healthcare
Life Sciences), centrifuged at 112 × g for 5 min at 37°C, and
resuspended in PBS. Cells were stained using the Annexin
V-Fluorescein Isothiocyanate Apoptosis Detection kit (BD
Biosciences, Franklin Lakes, NJ, USA), and propidium iodide (BD
Biosciences) according to the manufacturer's instructions. The
apoptosis was analyzed using flow cytometry (FC 500 MPL; Beckman
Coulter, Inc.).
ROS-generation assay
The intracellular ROS production levels were
determined using a spectrofluorimetric method, H2DCFDA (Beyotime
Institute of Biotechnology, Haimen, China) assay. R- and R+ cells
were grown to a number of 107 and exposed to hypoxic conditions for
24 and 48 h. Cells were then incubated with DCHF-DA (20 mM) for 20
min at 37°C in a dark room. Subsequently, the cells were harvested
in a trypsin-EDTA acid solution. Cell suspensions were centrifuged
at 175 × g for 5 min at 37°C, then the supernatant was removed. The
intensity of DCHF-DA fluorescence was measured and calculated by
flow cytometry analysis (FC 500 MPL; Beckman Coulter, Inc.).
Statistical analysis
All experiments were performed in triplicate. Data
were expressed as the mean ± standard deviation. Statistical
analysis was performed using the paired Student's t-test with SPSS
software, version 22.0 (IBM SPSS, Armonk, NY, USA) considering the
variances unequal. P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of IGF-1R on hypoxia-induced
apoptosis
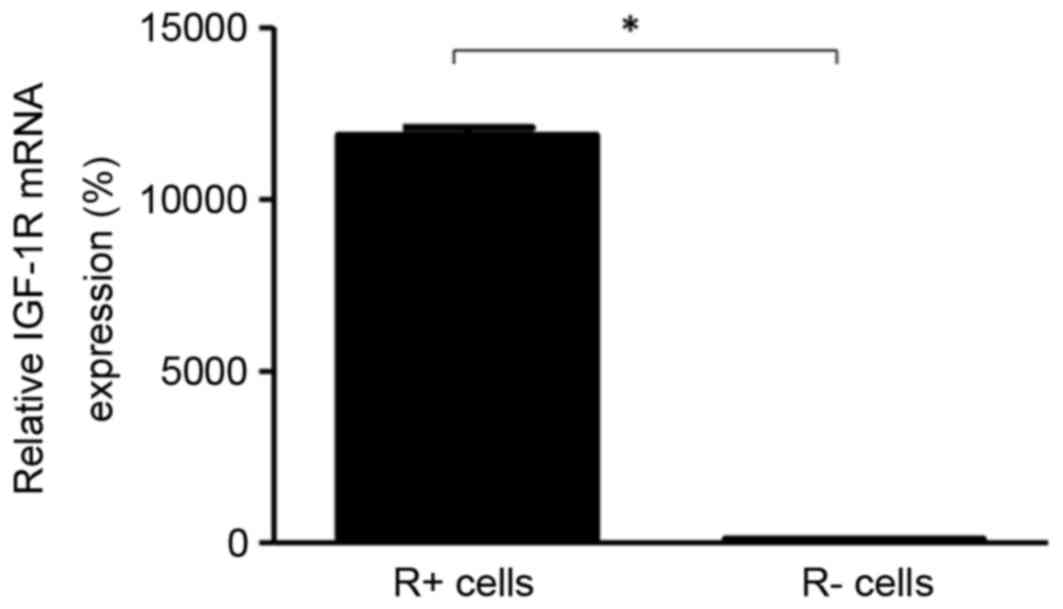
R+ cells exhibited significantly higher levels of
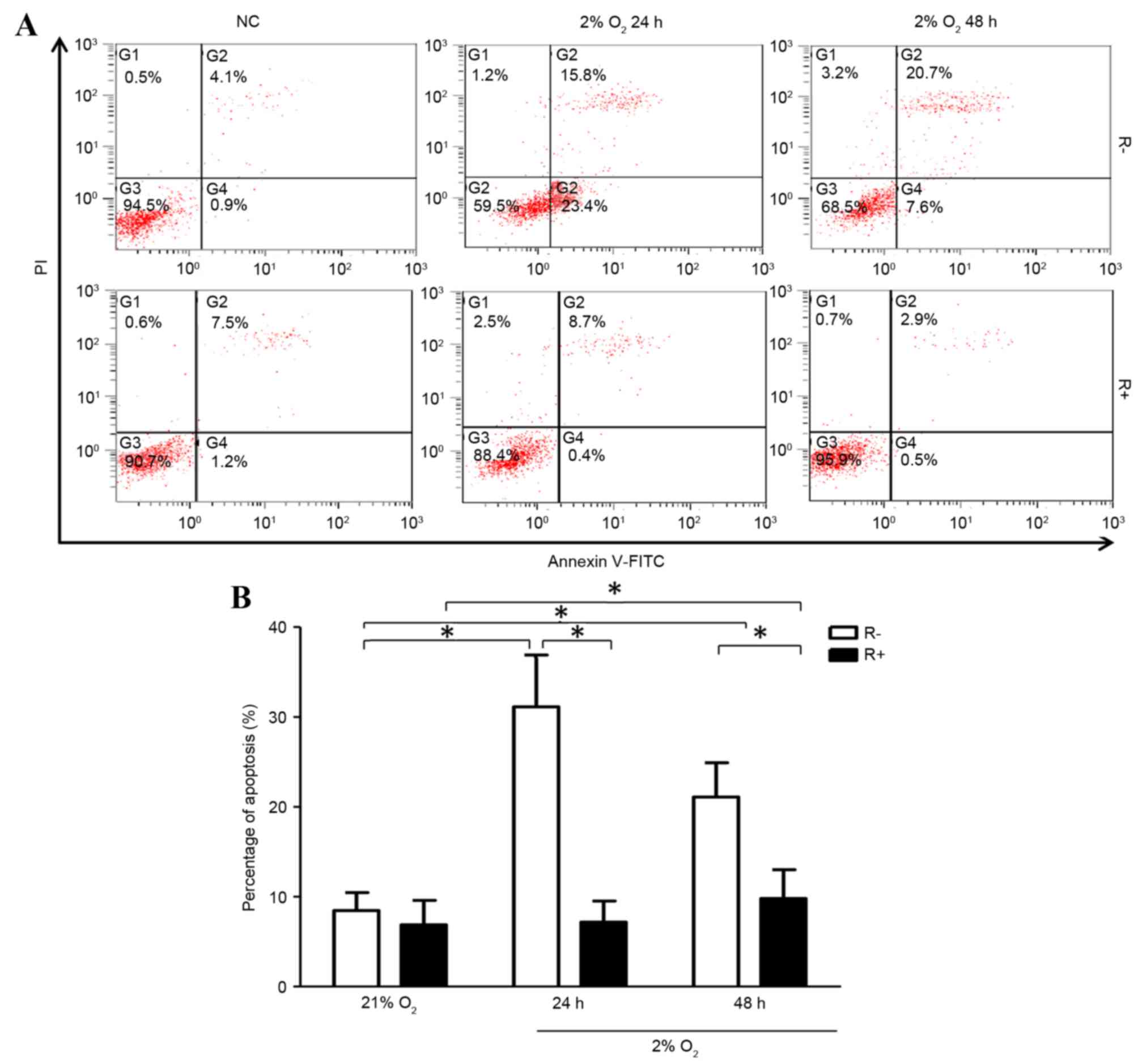
mRNA expression compared with that of R- cells (Fig. 1). Following treatment of hypoxia
for 24 or 48 h, R+ cells and R- cells all exhibited increased
apoptotic ratios compared with that of normoxic treatment,
respectively. Further analysis showed hypoxia induced more
apoptosis in R- cells compared with that of R+ cells under hypoxic
conditions (Fig. 2).
Effects of IGF-1R on hypoxia-induced
ROS production
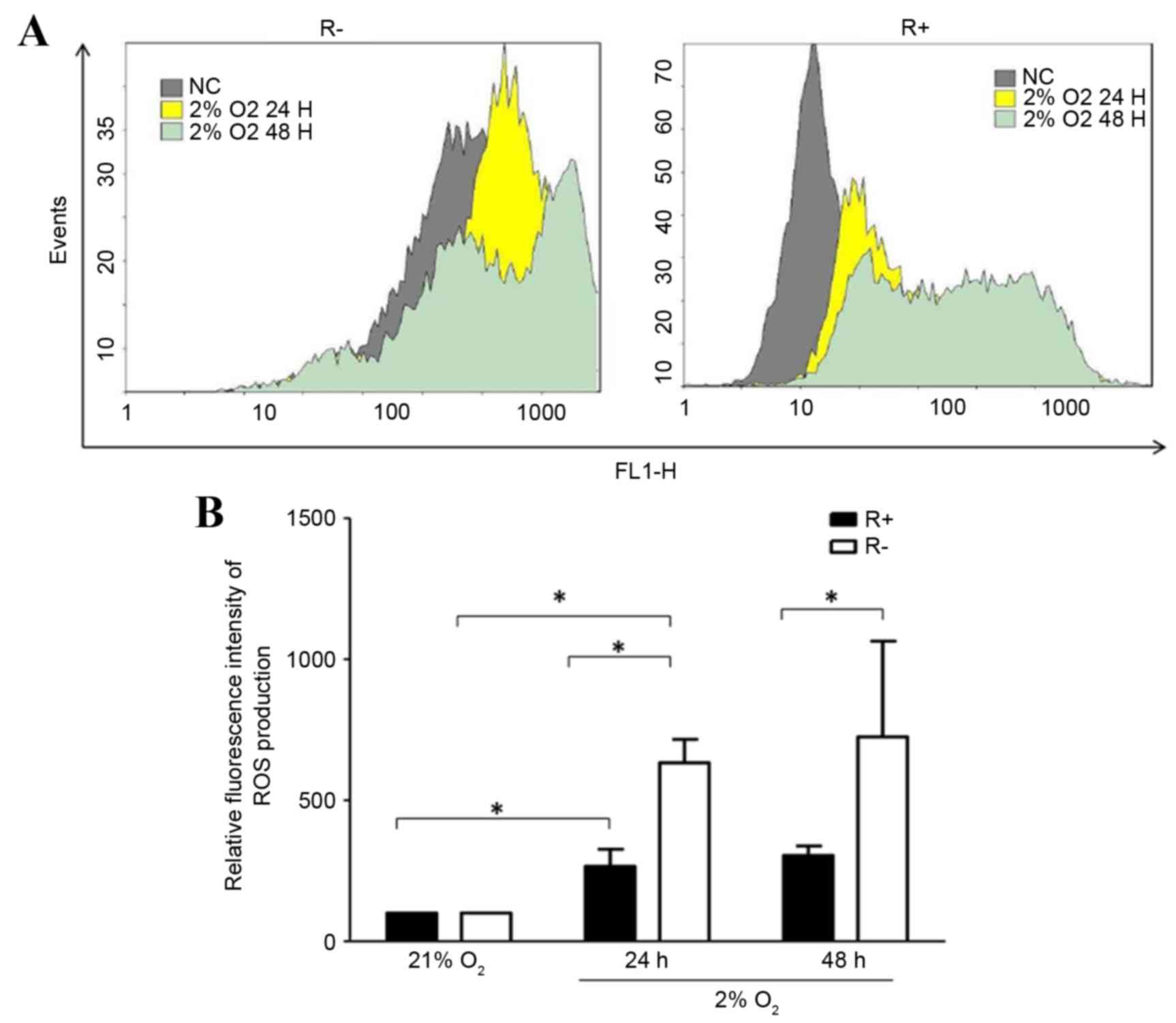
To investigate whether IGF-1R alters cellular ROS
levels under hypoxic conditions, ROS production in R- and R+ cells
under hypoxic conditions for 24 and 48 h was examined. As shown in
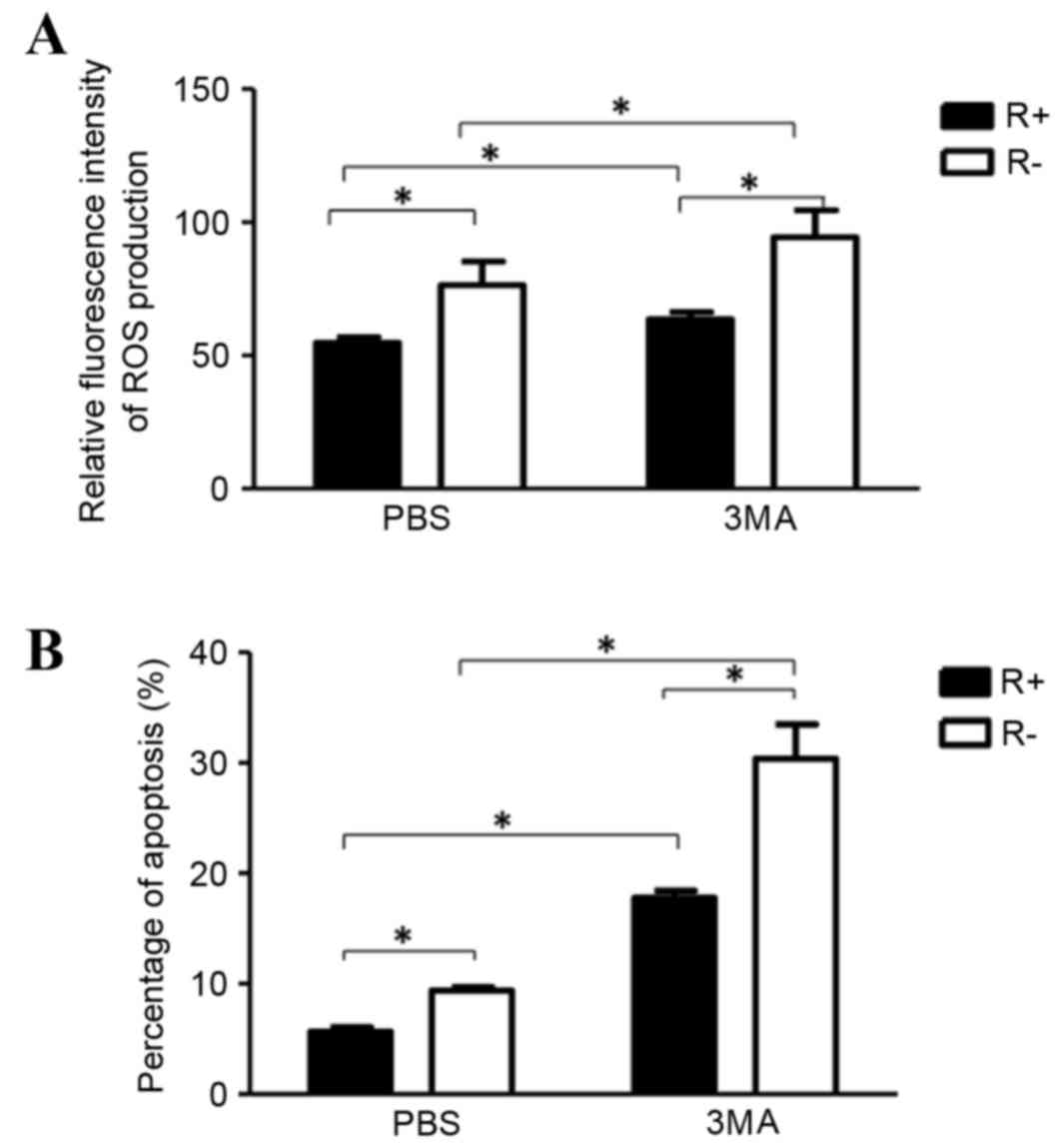
Fig. 3, ROS production levels in
R- and R+ cells was increased significantly under hypoxic
conditions compared with those under normoxic conditions.
Additionally, R+ cells produced significantly lower ROS levels
under hypoxic conditions. Under hypoxic conditions, the ROS levels
were significantly higher in R- cells than in R+ cells (Fig. 3).
Effects of IGF-1R on hypoxia-induced
autophagy
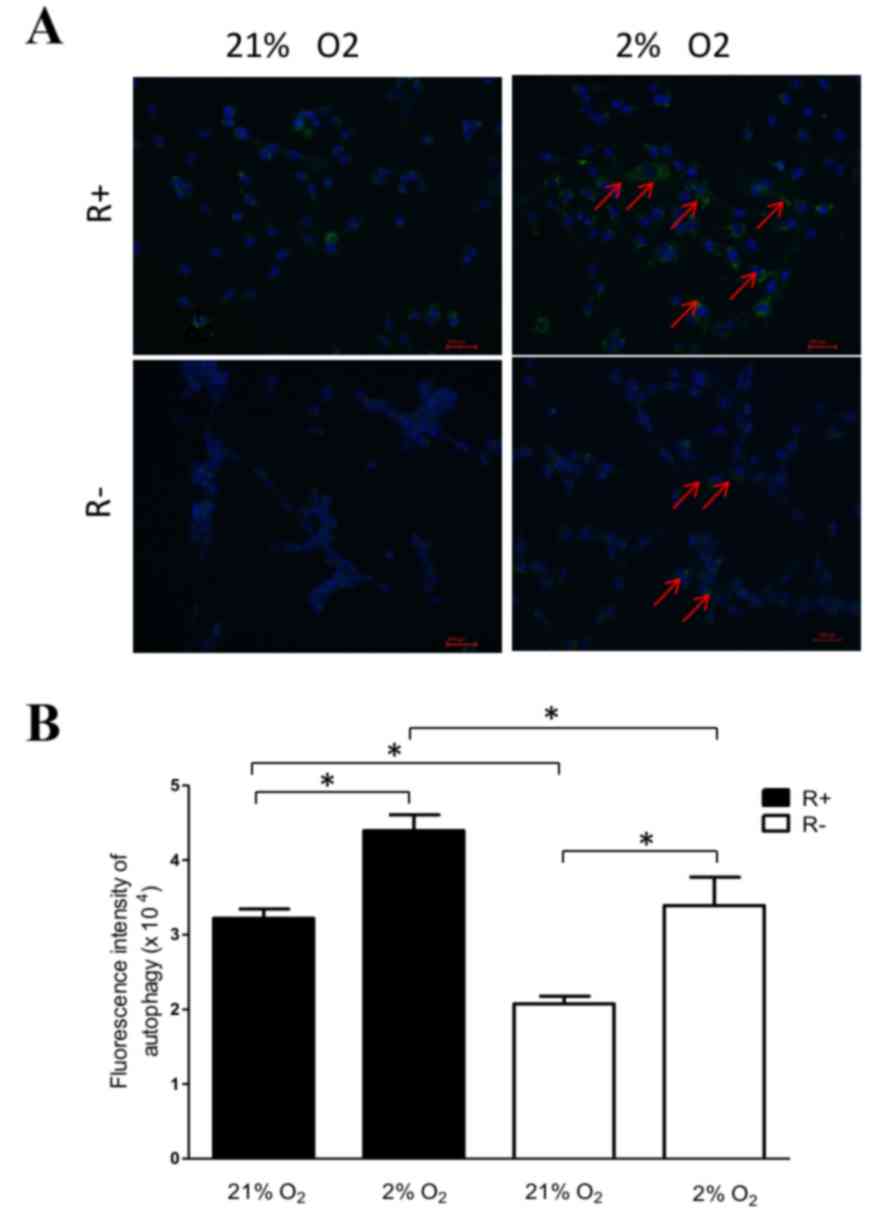
As presented in Fig.
4, hypoxia increased the presence of autophagosome in all R-
and R+ cells compared with that of normoxic treatment. In addition,
R+ cells exhibited higher levels of autophagy when compared with
R− cells under normoxic and hypoxic conditions, which
suggests that IGF-1R may promote autophagy.
Role of cell autophagy in ROS
production and apoptosis under hypoxic conditions
ROS production and apoptosis were higher in R- cells
compared with those in R+ cells. Further investigation indicated
that ROS production and apoptosis were increased after autophagy
inhibition (3MA) treatment in all R+ cells and R- cells compared
with those without autophagy inhibition (3MA) treatment (Fig. 5). These suggested that autophagy
may serve a protective role from ROS production and apoptosis.
Role of the PI3K/Akt/mTOR signaling
pathway in IGF-1R-induced autophagy under hypoxic conditions
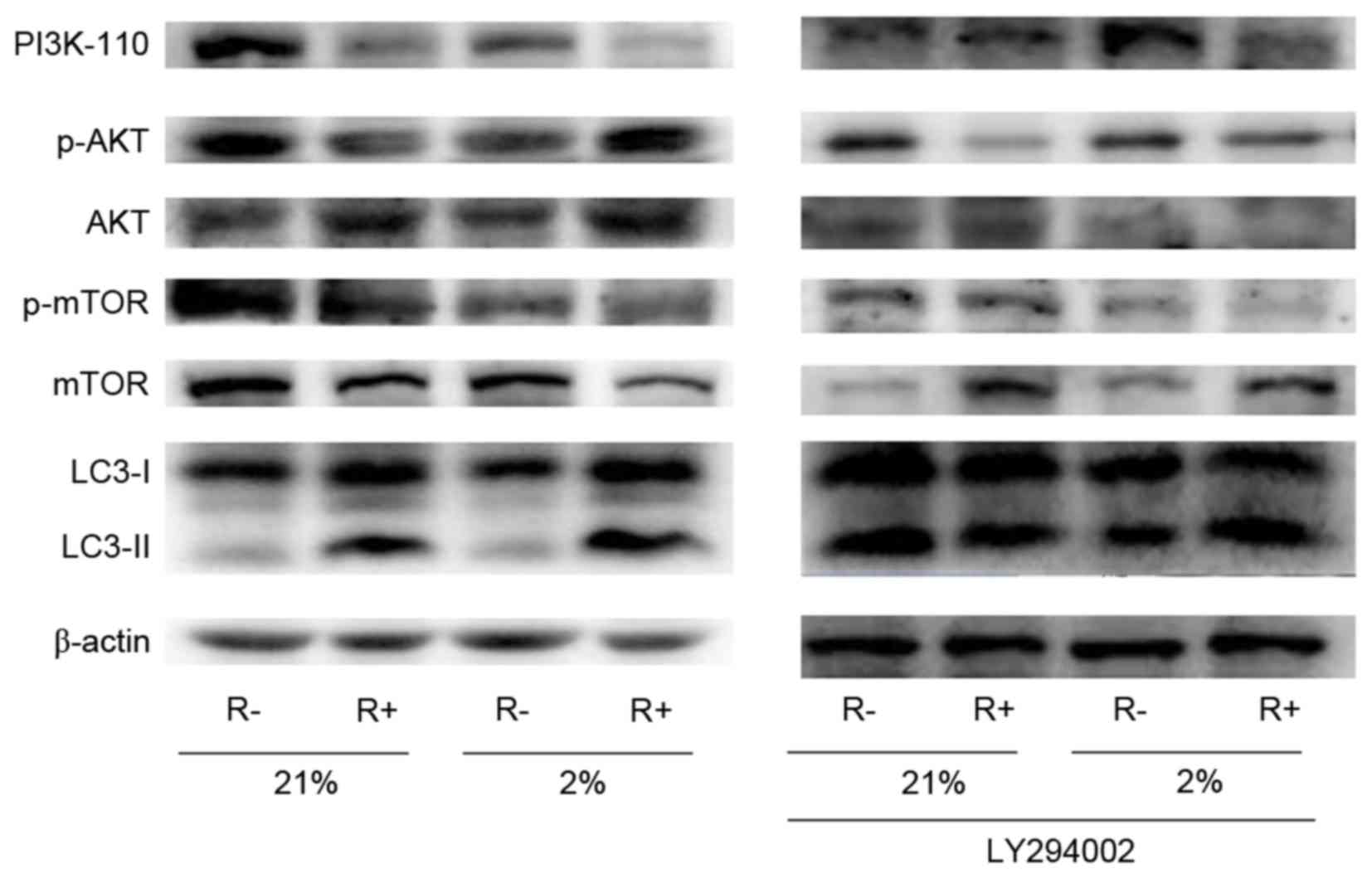
As shown in Fig. 6,
the R+ cells displayed lower expression levels of PI3K-110 and
mTOR, and higher expression of LC3-II under normoxic and hypoxic
conditions relative to R- cells. Further study indicated that p-AKT
was decreased in R+ cells compared with that of R- cells under
normoxic conditions, however was increased significantly in R+
cells following hypoxia treatment. When treated with the
PI3K/Akt/mTOR inhibitor LY294002, R+ and R- cells displayed lower
levels of PI3K/p-Akt/p-mTOR and high LC3-II expression. These
results indicated that the PI3K/Akt/mTOR signaling pathway may be
involved in autophagy under normoxic and hypoxic conditions, which
may be closely associated with IGF-1R.
Discussion
Hypoxia is a toxic factor that induces cell death
through mitochondrial dysfunction, which is primarily caused by the
production of ROS under hypoxic conditions (23,24).
Various pathological conditions, including myocardial I/R, stroke,
cancer and irradiation, lead to tissue hypoxia, which alter
biological characteristics, including the production of ROS,
autophagy and apoptosis. The results of the present study,
demonstrated that low concentrations of oxygen induce ROS
production and apoptosis in mouse embryonic fibroblasts. In
addition, overexpression of IGF-1R may serve a protective role in
cell death under hypoxic conditions through altering autophagy
levels and ROS production.
The role of IGF-1R has received extensive attention
due to its protective role in the neuronal system. It protects
neural cells from loss and infarcted volume, and increases glial
proliferation in the brain in a cerebral ischemia model (25–28).
In addition, it serves a role in protecting against renal
fibrosis-associated kidney disease, intestinal I/R injury and
myocardial ischemia (2,3,14).
Autophagy is an intracellular lysosomal degradation process that
maintains cellular homeostasis via the degradation and recycling of
long-lived proteins and intracellular aggregates as well as damaged
organelles, in order to generate small reusable molecules (29,30).
It is essential for the promotion of cell survival in stress
conditions, such as starvation, oxidative stress, and hypoxia;
however, unregulated autophagy induces progressive consumption of
cellular contents and results in autophagic cell death (31). Autophagy serves a protective role
in cell survival in cells through the reduction of ROS. Inhibition
of autophagy through chloroquine administration, or via prevention
of K63 ubiquitination increases the formation of ROS (32,33).
Hypoxia is a known inducer of autophagy (34). The present study demonstrated that
the induction of autophagy and ROS production under hypoxic
conditions, and autophagy inhibition by 3MA, led to a higher level
of ROS production and cell apoptosis in R+ and
R− cells, which is in agreement with a previous study
(35).
The signaling pathways involved in the progression
of autophagy are complex, and the PI3K/Akt pathway has been widely
studied (36,37). The anti-apoptotic effects of IGF-1
are mediated by IGF-1R, together with the subsequent activation of
PI3K/Akt/mTOR and other signaling pathways (38). However, the role of IGF-1 in
autophagy remains unknown. An inhibitory effect of IGF-1 on
autophagy has been observed in rat cardiomyocytes (39), human fibroblasts (40), and vascular cells from patients
with atherosclerotic lesions (41). These effects are associated with
rescuing mitochondrial metabolism and controlling the potentially
harmful autophagic response. By contrast, IGF-1 promotes autophagy
in H9c2 cell lines (42), HeLa
cells (43) and Purkinje neurons
(44). The depletion of IGF-1R
inhibits mTORC2 and reduces the activation of protein kinase C,
which decreases the rate of clathrin-dependent endocytosis and
impacts autophagosome-precursor formation (43). Furthermore, IGF-1 prevents the
accumulation of autophagic vesicles and cell death by increasing
the rate of autophagosome-to-lysosome fusion and degradation,
thereby contributing to cell survival (44).
The current study investigated the effects of the
IGF-1/PI3K/Akt/mTOR signaling pathway in the progression of
autophagy in mouse embryonic fibroblasts. The results indicated
that IGF-1R-overexpressing cells exhibit lower expression of
PI3K-110, p-Akt, p-mTOR/mTOR and higher expression levels of LC3-II
under normoxic and hypoxic conditions. When treated with the
PI3K/Akt/mTOR inhibitor LY294002, these cell types exhibited lower
PI3K/Akt/mTOR signaling activation and higher autophagy levels,
which indicated that IGF-1R-induced cell survival during hypoxia,
which may be dependent on autophagy initiation by suppressing the
PI3K/Akt/mTOR-signaling pathway.
The present study demonstrated that overexpression
of IGF-1R is correlated with reduced ROS production, increased
autophagy and cell viability under hypoxic conditions. It is
possible that, binding of IGF-1 to IGF-1R suppressed the
PI3K/Akt/mTOR signaling pathway, which promoted autophagy and
scavenging of redundant cellular ROS during hypoxia. These results
reveal novel mechanisms for IGF-1R-associated cell survival, which
may be good to know I/R- and H/I associated hypoxia-agravated
normal tissue injury well.
Acknowledgements
The current study was supported by the Scientific
Research Foundation for the Returned Overseas Chinese Scholars from
the China State Education Ministry (No. N130204), the National
Natural Science Foundation of China (Nos. 81202148 and 31370838)
and the Shanghai Pujiang Program (No. 13P1401600).
Glossary
Abbreviations
Abbreviations:
|
Akt
|
threonine protein kinase B
|
|
DCHF-DA
|
dichloro-dihydro-fluorescein
diacetate
|
|
H2DCFDA
|
2′, 7′-dichlorodihydrofluorescein
diacetate
|
|
IGF-1R
|
insulin-like growth factor-1
receptor
|
|
3MA
|
3-methyladenine
|
|
mTOR
|
mammalian target of rapamycin
|
|
PI3K
|
phosphatidylinositol 3-kinase
|
|
ROS
|
reactive oxygen species
|
References
|
1
|
Liu W, D'Ercole JA and Ye P: Blunting type
1 insulin-like growth factor receptor expression exacerbates
neuronal apoptosis following hypoxic/ischemic injury. BMC Neurosci.
12:642011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Du L, Yu Y, Ma H, Lu X, Ma L, Jin Y and
Zhang H: Hypoxia enhances protective effect of placental-derived
mesenchymal stem cells on damaged intestinal epithelial cells by
promoting secretion of insulin-like growth factor-1. Int J Mol Sci.
15:1983–2002. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li HX, Zhou YF, Zhao X, Jiang B and Yang
XJ: GATA-4 protects against hypoxia-induced cardiomyocyte injury:
Effects on mitochondrial membrane potential. Can J Physiol
Pharmacol. 92:669–678. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Caldwell CC, Tschoep J and Lentsch AB:
Lymphocyte function during hepatic ischemia/reperfusion injury. J
Leukoc Biol. 82:457–464. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ildefonso JA and Arias-Diaz J:
Pathophysiology of liver ischemia-reperfusion injury. Cir Esp.
87:202–209. 2010.(In Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vujaskovic Z, Anscher MS, Feng QF, Rabbani
ZN, Amin K, Samulski TS, Dewhirst MW and Haroon ZA:
Radiation-induced hypoxia may perpetuate late normal tissue injury.
Int J Radiat Oncol Biol Phys. 50:851–855. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vujaskovic Z, Marks LB and Anscher MS: The
physical parameters and molecular events associated with
radiation-induced lung toxicity. Semin Radiat Oncol. 10:296–307.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Luo Y, Liu X, Zheng Q, Wan X, Ouyang S,
Yin Y, Sui X, Liu J and Yang X: Hydrogen sulfide prevents
hypoxia-induced apoptosis via inhibition of an H2O2-activated
calcium signaling pathway in mouse hippocampal neurons. Biochem
Biophys Res Commun. 425:473–477. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim BM and Chung HW: Hypoxia/reoxygenation
induces apoptosis through a ROS-mediated caspase-8/Bid/Bax pathway
in human lymphocytes. Biochem Biophys Res Commun. 363:745–750.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Peng C, Rao W, Zhang L, Wang K, Hui H,
Wang L, Su N, Luo P, Hao YL, Tu Y, et al: Mitofusin 2 ameliorates
hypoxia-induced apoptosis via mitochondrial function and signaling
pathways. Int J Biochem Cell Biol. 69:29–40. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kuo CY, Chiu YC, Lee AY and Hwang TL:
Mitochondrial Lon protease controls ROS-dependent apoptosis in
cardiomyocyte under hypoxia. Mitochondrion. 23:7–16. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Laron Z: Insulin-like growth factor 1
(IGF-1): A growth hormone. Mol Pathol. 54:311–316. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chisalita SI and Arnqvist HJ: Insulin-like
growth factor I receptors are more abundant than insulin receptors
in human micro- and macrovascular endothelial cells. Am J Physiol
Endocrinol Metab. 286:E896–E901. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liang M, Woodard LE, Liang A, Luo J,
Wilson MH, Mitch WE and Cheng J: Protective role of insulin-like
growth factor-1 receptor in endothelial cells against unilateral
ureteral obstruction-induced renal fibrosis. Am J Pathol.
185:1234–1250. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Custodio RJ, do Carmo Custodio VI,
Scrideli CA, Milani SL Sader, Cervi MC, Cupo P and Martinelli CE
Jr: Impact of hypoxia on IGF-I, IGF-II, IGFBP-3, ALS and IGFBP-1
regulation and on IGF1R gene expression in children. Growth Horm
IGF Res. 22:186–191. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Morgan BL and Chao CR: The effects of
hypoxia on growth cones in the ovine fetal brain. J Matern Fetal
Neonatal Med. 16:55–59. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Heck S, Lezoualc'h F, Engert S and Behl C:
Insulin-like growth factor-1-mediated neuroprotection against
oxidative stress is associated with activation of nuclear factor
kappaB. J Biol Chem. 274:9828–9835. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Baregamian N, Song J, Jeschke MG, Evers BM
and Chung DH: IGF-1 protects intestinal epithelial cells from
oxidative stress-induced apoptosis. J Surg Res. 136:31–37. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Maldonado C, Cea P, Adasme T, Collao A,
Díaz-Araya G, Chiong M and Lavandero S: IGF-1 protects cardiac
myocytes from hyperosmotic stress-induced apoptosis via CREB.
Biochem Biophys Res Commun. 336:1112–1118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yu D, Watanabe H, Shibuya H and Miura M:
Redundancy of radioresistant signaling pathways originating from
insulin-like growth factor I receptor. J Biol Chem. 278:6702–6709.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu D, Shibuya H and Miura M: Roles of the
insulin-like growth factor I receptor C-terminus in cellular
radioresistance. Biochem Biophys Res Commun. 311:174–178. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Martin M, Lefaix J and Delanian S:
TGF-beta1 and radiation fibrosis: A master switch and a specific
therapeutic target? Int J Radiat Oncol Biol Phys. 47:277–290. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Murphy MP: How mitochondria produce
reactive oxygen species. Biochem J. 417:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lopez-Lopez C, LeRoith D and Torres-Aleman
I: Insulin-like growth factor I is required for vessel remodeling
in the adult brain. Proc Natl Acad Sci USA. 101:9833–9838. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Conti E, Carrozza C, Capoluongo E, Volpe
M, Crea F, Zuppi C and Andreotti F: Insulin-like growth factor-1 as
a vascular protective factor. Circulation. 110:2260–2265. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Adams MM, Forbes M Elizabeth, Linville M
Constance, Riddle DR, Sonntag WE and Brunso-Bechtold JK: Stability
of local brain levels of insulin-like growth factor-I in two
well-characterized models of decreased plasma IGF-I. Growth
Factors. 27:181–188. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Carro E, Spuch C, Trejo JL, Antequera D
and Torres-Aleman I: Choroid plexus megalin is involved in
neuroprotection by serum insulin-like growth factor I. J Neurosci.
25:10884–10893. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Avivar-valderas A, Bobrovnikova-Marjon E,
Diehl J Alan, Bardeesy N, Debnath J and Aguirre-Ghiso JA:
Regulation of autophagy during ECM detachment is linked to a
selective inhi- bition of mTORC1 by PERK. Oncogene. 32:4932–4940.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu B, Wen X and Cheng Y: Survival or
death: Disequilibrating the oncogenic and tumor suppressive
autophagy in cancer. Cell Death Dis. 4:e8922013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: Apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rouschop KM, Ramaekers CH, Schaaf MB,
Keulers TG, Savelkouls KG, Lambin P, Koritzinsky M and Wouters BG:
Autophagy is required during cycling hypoxia to lower production of
reactive oxygen species. Radiother Oncol. 92:411–416. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Farombi EO: Genotoxicity of chloroquine in
rat liver cells: Protective role of free radical scavengers. Cell
Biol Toxicol. 22:159–167. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sun Y, Xing X, Liu Q, Wang Z, Xin Y, Zhang
P, Hu C and Liu Y: Hypoxia-induced autophagy reduces
radiosensitivity by the HIF-1α/miR-210/Bcl-2 pathway in colon
cancer cells. Int J Oncol. 46:750–756. 2015.PubMed/NCBI
|
|
35
|
Liu Q, Sun Y, Lv Y, Le Z, Xin Y, Zhang P
and Liu Y: TERT alleviates irradiation-induced late rectal injury
by reducing hypoxia-induced ROS levels through the activation of
NF-κB and autophagy. Int J Mol Med. 38:785–793. 2016.PubMed/NCBI
|
|
36
|
Shao X, Lai D, Zhang L and Xu H: Induction
of autophagy and apoptosis via PI3K/AKT/TOR pathways by
azadirachtin a in spodoptera litura cells. Sci Rep. 6:354822016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fan XJ, Wang Y, Wang L and Zhu M:
Salidroside induces apoptosis and autophagy in human colorectal
cancer cells through inhibition of PI3K/Akt/mTOR pathway. Oncol
Rep. 36:3559–3567. 2016.PubMed/NCBI
|
|
38
|
Opgaard O Saetrum and Wang PH: IGF-I is a
matter of heart. Growth Horm IGF Res. 15:89–94. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Troncoso R, Vicencio JM, Parra V,
Nemchenko A, Kawashima Y, Del Campo A, Toro B, Battiprolu PK,
Aranguiz P, Chiong M, et al: Energy-preserving effects of IGF-1
antagonize starvation-induced cardiac autophagy. Cardiovasc Res.
93:320–329. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bitto A, Lerner C, Torres C, Roell M,
Malaguti M, Perez V, Lorenzini A, Hrelia S, Ikeno Y, Matzko ME, et
al: Long-term IGF-I exposure decreases autophagy and cell
viability. PLoS One. 5:e125922010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jia G, Cheng G, Gangahar DM and Agrawal
DK: Insulin-like growth factor-1 and TNF-alpha regulate autophagy
through c-jun N-terminal kinase and Akt pathways in human
atherosclerotic vascular smooth cells. Immunol Cell Biol.
84:448–454. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Aki T, Yamaguchi K, Fujimiya T and
Mizukami Y: Phosphoinositide 3-kinase accelerates autophagic cell
death during glucose deprivation in the rat cardiomyocyte-derived
cell line H9c2. Oncogene. 22:8529–8535. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Renna M, Bento CF, Fleming A, Menzies FM,
Siddiqi FH, Ravikumar B, Puri M, Garcia-Arencibia M, Sadiq O,
Corrochano S, et al: IGF-1 receptor antagonism inhibits autophagy.
Hum Mol Genet. 22:4528–4544. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bains M, Florez-McClure ML and Heidenreich
KA: Insulin-like growth factor-I prevents the accumulation of
autophagic vesicles and cell death in Purkinje neurons by
increasing the rate of autophagosome-to-lysosome fusion and
degradation. J Biol Chem. 284:20398–20407. 2009. View Article : Google Scholar : PubMed/NCBI
|