Introduction
Crohn's disease (CD) is a chronic and relapsing
inflammatory disease, which can affect any part of the
gastrointestinal tract. Over the last few decades, CD has become
progressively common in several developed countries, affecting
almost one in 200 individuals, and its incidence and prevalence are
increasing rapidly in developing countries (1). Patients with CD usually present with
abdominal pain and severe diarrhea, accompanied with fever and
serious weight loss. The fluctuating course of CD readily leads to
patients experiencing stress and other psychosocial problems,
significantly affecting quality of life in terms of health. Despite
the wide use of immunosuppressive and anti-tumor necrosis factor α
(anti-TNFα) therapies, >50% of patients require surgery within
10 years of diagnosis (2).
Therefore, additional investigations into the etiology and
molecular pathogenesis are required to improve clinical management
of this disease.
The rapid development of microarray technology has
provided an innovative approach for examining the biological
mechanism of diseases. Previous studies based on genome-wide
expression analyses have found novel biomarkers of inflammatory
bowel disease and several other diseases (3–5). As
this high-throughput technology can assist in revealing the
etiology and pathogenesis underlying the disease course, the
present study screened differentially expressed genes between colon
tissues of patients with CD and control samples with microarray
data obtained from the Gene Expression Omnibus (GEO) database. The
significant functions and signaling pathways in which these genes
are enriched were then identified, and the microRNAs (miRNAs)
regulating these genes were predicted. The aim of these
investigations was to obtain an improved understand of the
pathogenesis, and provide novel diagnostic and therapeutic
approaches for CD.
Materials and methods
Data resources and preprocessing
A total of seven microarray profiles of CD, which
were constructed within the last 10 years, were obtained from the
GEO (http://www.ncbi.nim.nih.gov/geo/)
database (accession nos. GSE6731, GSE9686, GSE10616, GSE20881,
GSE26305, GSE36807 and GSE52746) (6). A total of 267 samples, including 116
normal control colon samples and 151 colon tissue samples from
patients with CD without TNFα therapy were included for the
analyses. Subsequently, the original data were converted into probe
expression measurements. The average value was applied for probes
matching one gene from the same profile, and an intersection of the
seven profiles was then performed, which obtained 7,793 common
genes. Normal distribution was used to standardize each profile, as
reported previously (7).
Screening of differentially expressed
genes
The differently expressed genes between the control
and CD sample groups were identified as previously reported
(8). In brief, a random variance
model t-test was performed using SPSS software (version, 20.0; IBM
SPSS, Armonk, NY, USA) to filter the differently expressed genes,
following which the false discovery rate (FDR) and significance
were calculated. Genes with P<0.05 and FDR<0.05 values were
considered to be significantly different.
Gene ontology (GO) analysis
Using data from the GO database (http://www.geneontology.org/), GO analysis was
performed to determine the function of the significantly enriched
differentially expressed genes, as reported previously (9). For classifying the GO categories, the
χ2 test and Fisher's exact test were performed using SPSS software
(version, 22.0; IBM SPSS). FDR<0.05 was used to correct for
multiple comparisons, and the GO terms with P<0.05 and
FDR<0.05 were considered significant. Subsequently, enrichment
values were calculated to select significant GO terms with the most
concrete description of function. A GO network was then formed
using Cytoscape v3.2.0 (http://cytoscape.org/) to summarize associated
interactions on a GO map.
Pathway enrichment analysis
To investigate the significant pathways in which the
differentially expressed genes were enriched, the Kyoto
Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg/) was used to perform a KEGG
analysis (10). As above, the χ2
test and Fisher's exact test were used to identify significant
pathways. In addition, the FDR, P-value and enrichment values were
calculated. P<0.05 and FDR<0.05 were regarded as the cutoff
for selecting significantly enriched pathways. Finally, Cytoscape
v3.2.0 was used to establish a path-net to outline the association
among these significant pathways.
miRNA-gene network
To investigate the regulatory miRNAs of these
differentially expressed genes, the miRNAs were predicted using
TargetScan (http://www.targetscan.org/) and miRDB (http://www.mirdb.org/), and the results of these two
databases were intersected to guarantee the accuracy of the
prediction (11,12). To elucidate the possible links
between the differentially expressed genes, a miRNA-gene network
was constructed using Cytoscape v3.2.0.
Results
Differentially expressed genes in CD,
compared with normal colon tissue
A total of 432 differentially expressed genes,
including 229 upregulated genes and 203 downregulated genes were
screened out in the CD samples, compared with the normal colon
tissues (Fig. 1). As shown in
Fig. 1, the top five upregulated
differentially expressed genes were matrix metallopeptidase (MMP)3,
chemokine (C-X-C motif) ligand 1 (CXCL1), MMP1, regenerating family
member 3α (REG3A) and CXCL9, whereas the downregulated genes were
glutathione S-transferase α1 (GSTA1), carbonic anhydrase I (CA1),
ATP synthase, H+ transporting, mitochondrial Fo complex subunit B1
(ATP5F1), heat shock protein family B (small) member 3 (HSPB3) and
interleukin 2 (IL-2).
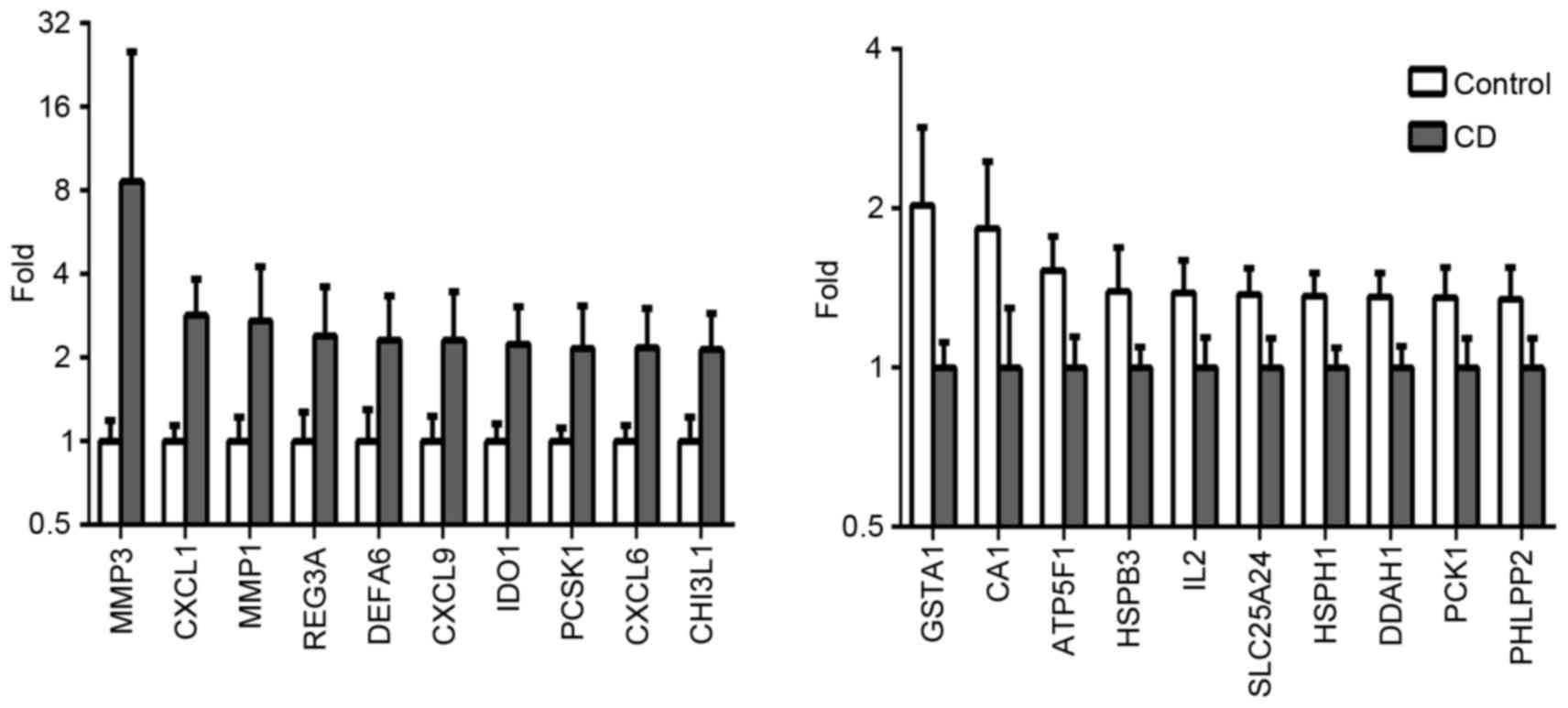 | Figure 1.Differentially expressed genes in CD.
Graphs show the top 10 genes most markedly upregulated and
downregulated in CD, compared with normal colon tissues. P<0.05;
FDR<0.05. Data are presented as the geometric mean with 95%CI.
CD, Crohn's disease. MMP3, matrix metallopeptidase 3; CXCL,
chemokine (C-X-C motif) ligand; REG3A, regenerating family member
3α; DEFA6, defensin α 6; IDO1, indoleamine 2,3-dioxygenase 1;
PCSK1, proprotein convertase subtilisin/kexin type 1; CHI3L1,
chitinase 3 like 1; GSTA1, glutathione S-transferase α1; CA1,
carbonic anhydrase I, ATP5F1, ATP synthase, H+
transporting, mitochondrial Fo complex subunit B1; HSPB3, heat
shock protein family B (small) member 3; IL-2, interleukin 2;
SLC25A24, solute carrier family 25 member 24; DDAH1,
dimethylarginine dimethylaminohydrolase 1; PKC1, protein kinase C1;
PHLPP2, PH domain and leucine rich repeat protein phosphatase
2. |
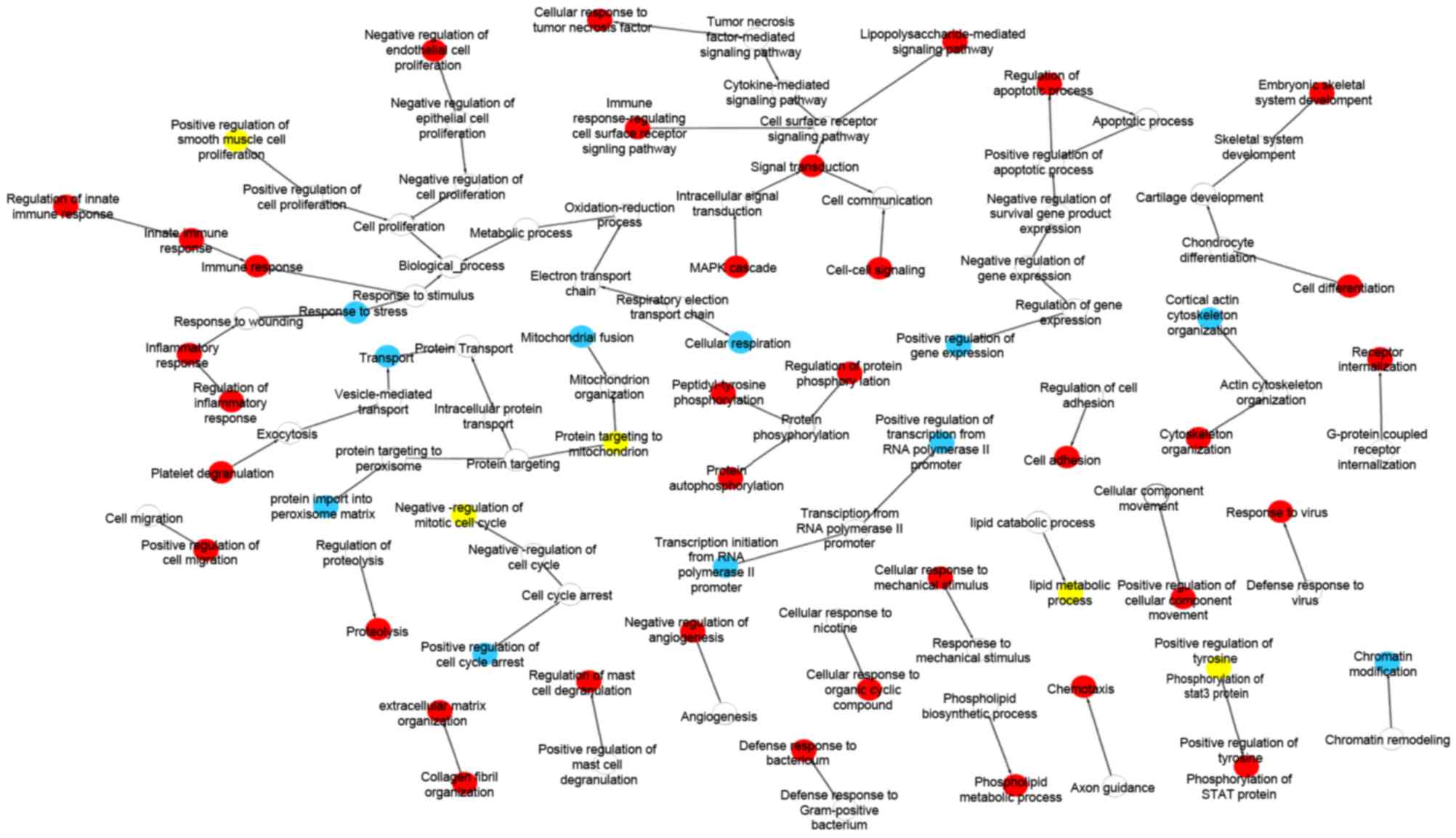
GO enrichment analysis
GO analysis was performed on these differentially
expressed genes to provide a preliminarily outlook on their
biological functions. As shown in Fig.
2, the upregulated genes were predominantly involved in
regulation of inflammatory response, innate immune response, mast
cell degranulation, cellular response to TNF and defense response
to bacterium, indicating that innate immune system and microbial
factors play an important role in inducing inflammation in CD. They
were also enriched in proteolysis, chemotaxis, cytoskeleton
organization and cell adhesion, and these functions enhance cell
migration. These genes were also associated with extracellular
matrix (ECM) organization, collagen fibril organization and
mitogen-activated protein kinase cascade. However, the upregulated
genes were found to be involved in the negative regulation of
endothelial cell proliferation and angiogenesis, which was in
contrast to the majority of previous studies and may require
further investigation (13,14).
The downregulated genes were significantly enriched
in the regulation of transcription, transport, response to stress,
cellular respiration, chromatin modification and several other
metabolic processes. In addition, the two groups of differentially
expressed genes were involved in regulating the tyrosine
phosphorylation of signal transducers and activators of
transcription 3 (STAT3) protein, the mitotic cell cycle, smooth
muscle cell proliferation and lipid metabolic process.
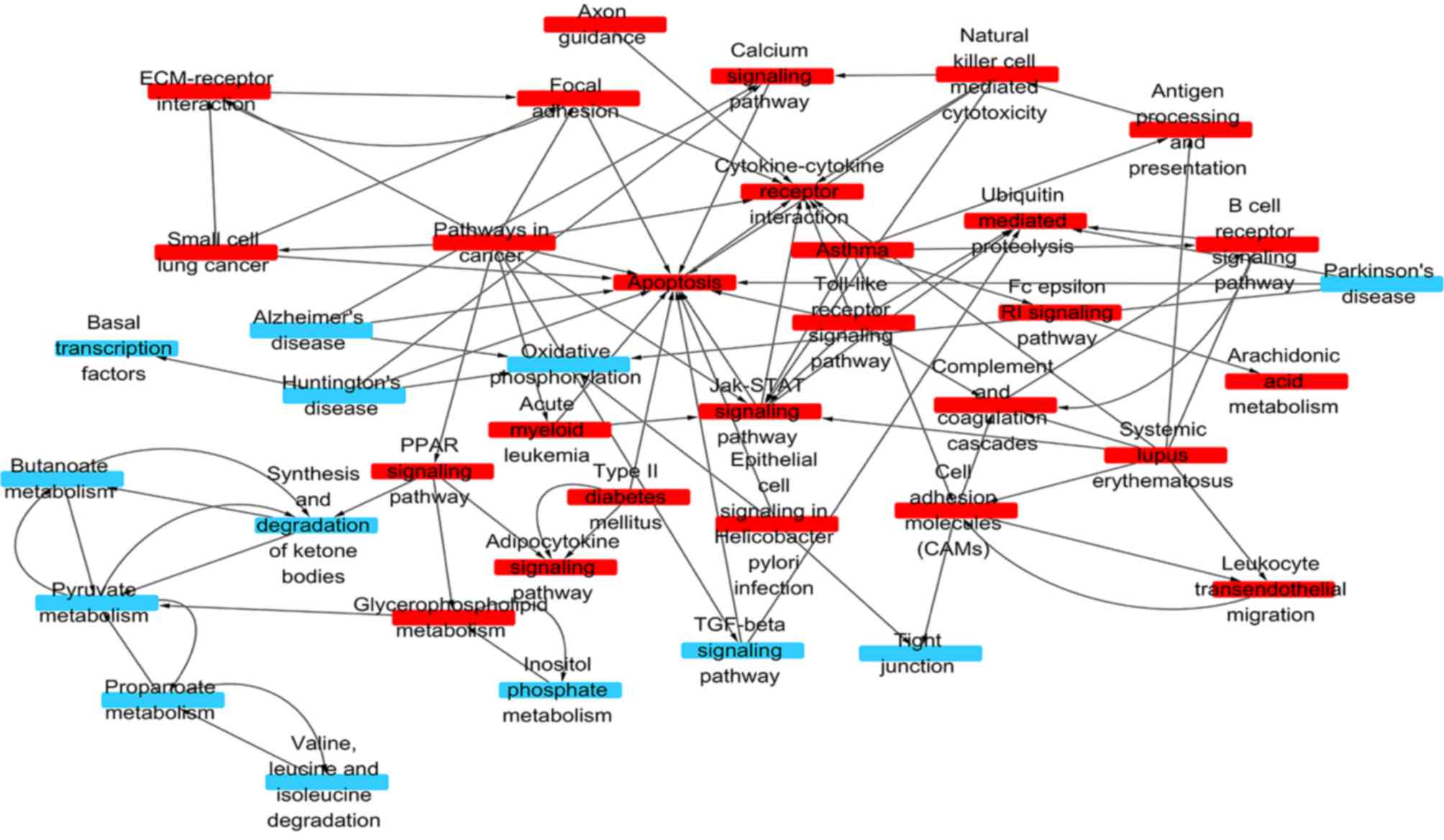
Signaling network analysis
As demonstrated in Fig.
3, the upregulated genes were primarily involved in focal
adhesion, cell adhesion molecules B cell receptor signaling
pathway, leukocyte transendothelial migration and natural killer
cell mediated cytotoxicity, consistent with the results of the GO
enrichment analyses. They were also associated with the Janus
kinase/STAT (JAK/STAT) signaling pathway, apoptosis, calcium
signaling pathway, cytokine-cytokine receptor interaction,
Toll-like receptor signaling pathway and the peroxisome
proliferator activated receptor. Other signaling pathways,
including phosphatidylinositol 3-kinase/Akt and nuclear factor kB,
were also upregulated, however, these were not statistically
significant and thus not shown.
The downregulated genes were enriched in the
transforming growth factor-β signaling pathway, tight junctions,
basal transcription factors, oxidative phosphorylation and several
other metabolic pathways.
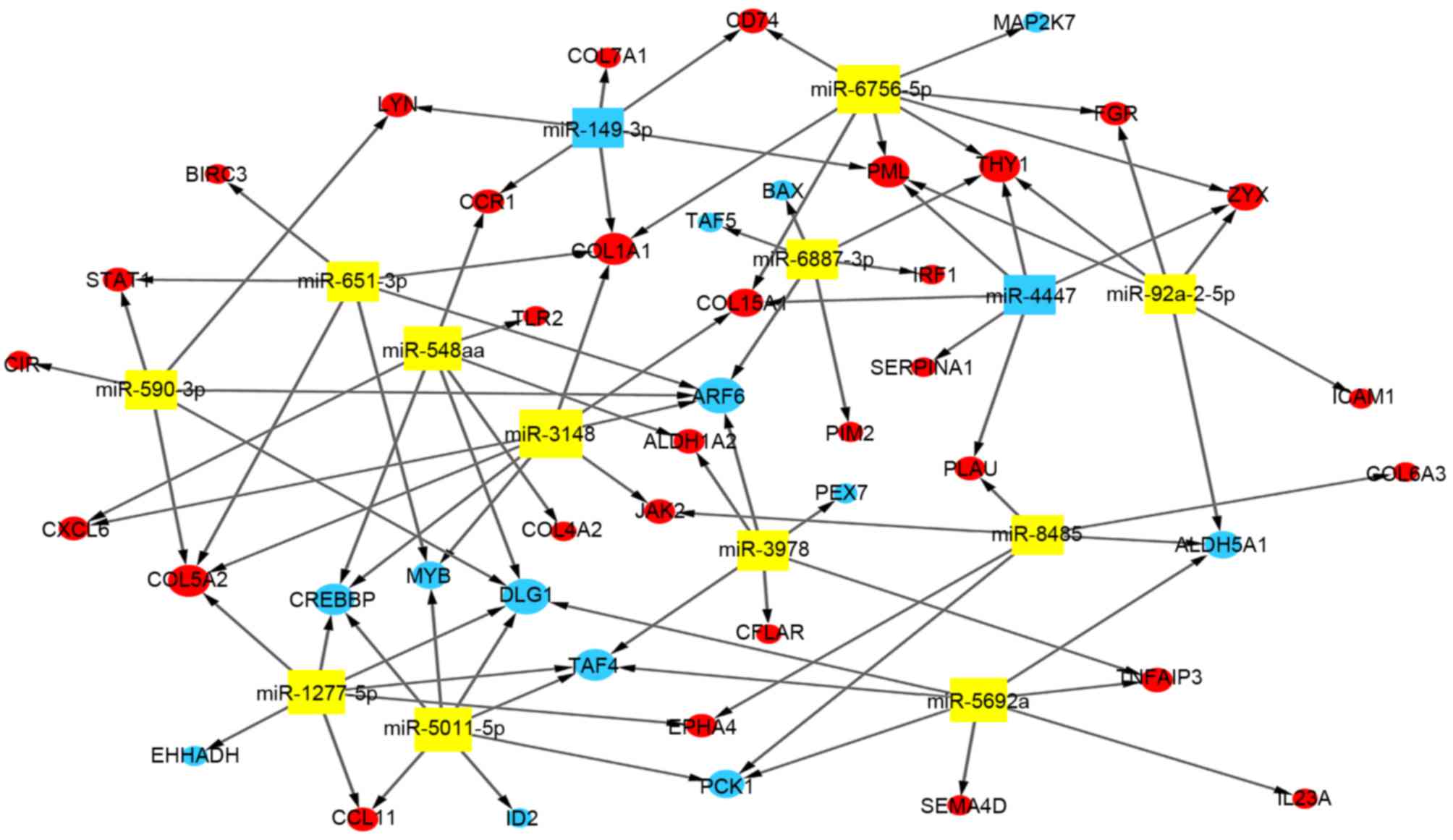
miRNA-gene network
Based on the regulated association between the
differentially expressed genes and miRNAs, a network of miRNA-genes
was constructed. As presented in Fig.
4, miR-3148 and miR-6756-5p regulated the majority of genes,
including JAK2 and collagen, type I, alpha 1 (COL1A1). miR-149-3p
and miR-4447 regulated upregulated genes only, whereas the
remaining miRNAs regulated upregulated and downregulated genes.
These miRNAs may be central in the development and progression of
CD.
Discussion
In the present study, the gene expression profiles
of CD and normal colon tissue samples were examined using
bioinformatics methods. It was found that 229 genes were
upregulated and 203 genes were downregulated in the colon tissues
of patients with CD, compared with those of normal controls. These
differentially expressed genes may serve as characteristic genes
closely associated with the diagnosis and treatment of CD,
particularly those ranked at the top of the list. Among these
genes, MMPs, including MMP1 and MMP3, are known to be
markedly upregulated in the inflamed intestinal mucosa of patients
with CD (15), which was in
accordance with the results of the present study. As the imbalance
of ECM components between the synthesis and breakdown in IBD can
result in progressive tissue destruction or the over-deposition of
collagens, finally causing ulcers, fistulas and fibrosis (16), MMPs are key in the intestinal
tissue remodeling process (17).
In addition, their upregulation may lead to patients with CD not
responding to anti-TNFα therapy (18). In terms of the downregulated genes,
GSTA1 may protect the cells from reactive oxygen species and
the products of peroxidation through glutathione peroxidase
activity. Restoring the expression of GSAT1 attenuates the
inflammatory response in the intestinal mucosa of CD (19). Therefore, GSAT1 may serve as
a novel therapeutic target and biomarker of prognosis in CD.
In the present study, GO analysis was performed to
further interpret which functions these differentially expressed
genes enriched. The results indicated that the functions included
regulation of inflammatory response, immune response, defense
response to bacterium and cellular response to tumor necrosis
factor, which was consistent with a previous study, which
identified CD is a complex inflammatory disease with the immune
response and microbiota involved in its pathogenesis (20). In addition, these differentially
expressed genes were significantly enriched in proteolysis, ECM
organization and collagen fibril organization, the functions of
which are important in the development of intestinal fibrosis and
CD-associated strictures (17). As
MMPs are involved in the degradation of the ECM, their potential
role in the pathogenesis of CD warrants further investigation. The
present study also showed that the functions of downregulated genes
comprised the response to stress and cellular respiration,
suggesting that oxidative stress is also involved in the etiology
of CD.
Signaling pathways of the target genes were assessed
using KEGG analysis. The results showed that several signaling
pathways, including the cytokine-cytokine receptor interaction and
the JAK/STAT signaling pathway, were associated with the occurrence
of CD. Previous studies have indicated that various cytokines,
including interleukins, regulate intracellular signaling through
inducing the JAK/STAT pathway, and then convert extracellular
stimuli into several cellular processes, for example, cell growth,
proliferation and differentiation (21,22).
In addition, the numerous pro-inflammatory cytokines secreted,
including IL-6 and TNF-α, by immune cells, including T cells and
neutrophils, may cause an inflammatory cascade by activating the
JAK/STAT pathway, resulting in a mucosal inflammatory response
(23). The findings indicated that
the JAK/STAT pathway may be essential in regulation of the immune
response and the pathogenesis of mucosal inflammation in CD, and
present a promising novel therapeutic strategy for CD.
Finally, a miRNA-gene network was constructed in the
present study to illustrate the internal association between the
differentially expressed genes and the miRNAs of the target genes.
Several miRNAs were found to be involved in the regulation of
differentially expressed genes and may be considered as crucial
regulators in the pathogenesis of CD. For example, miR-149 is
downregulated in multiple types of tumor (24–26),
and the overexpression of miR-149 can suppress cell migration, cell
proliferation and the cell cycle (25,27).
The downregulation of miR-149 can also lead to the high expression
of IL-6 (28), a pro-inflammatory
cytokine, which has been implicated in the pathogenesis of CD
(29). Therefore, the role of
miR-149 in CD requires further investigation, particularly as the
function of miR-149 in CD has not been investigated.
In conclusion, the present study identified a number
of differentially expressed genes, some of which may be important
in the diagnosis and treatment of CD. Furthermore, using GO and
KEGG analyses, combined with the construction of an miRNA-gene
network, important gene functions, pathways and miRNAs, were
identified, which may provide novel insights on the molecular
mechanism and treatment of CD. However, further evidence from
independent experimental data is required to confirm these
results.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant nos. 81401875 and 81100263;
http://www.nsfc.gov.cn/).
References
|
1
|
Molodecky NA, Soon IS, Rabi DM, Ghali WA,
Ferris M, Chernoff G, Benchimol EI, Panaccione R, Ghosh S, Barkema
HW and Kaplan GG: Increasing incidence and prevalence of the
inflammatory bowel diseases with time, based on systematic review.
Gastroenterology. 142:46–54.e42. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Frolkis AD, Dykeman J, Negrón ME, Debruyn
J, Jette N, Fiest KM, Frolkis T, Barkema HW, Rioux KP, Panaccione
R, et al: Risk of surgery for inflammatory bowel diseases has
decreased over time: A systematic review and meta-analysis of
population-based studies. Gastroenterology. 145:996–1006. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
van Limbergen J, Philpott D and Griffiths
AM: Genetic profiling in inflammatory bowel disease: From
association to bedside. Gastroenterology. 141:1566–1571.e1. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen L, Zhuo D, Chen J and Yuan H:
Screening feature genes of lung carcinoma with DNA microarray
analysis. Int J Clin Exp Med. 8:12161–12171. 2015.PubMed/NCBI
|
|
5
|
Zhan C, Yan L, Wang L, Jiang W, Zhang Y,
Xi J, Jin Y, Chen L, Shi Y, Lin Z and Wang Q: Landscape of
expression profiles in esophageal carcinoma by the cancer genome
atlas data. Dis Esophagus. 29:920–928. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res. 41(Database issue):
D991–D995. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Montero-Melendez T, Llor X,
García-Planella E, Perretti M and Suárez A: Identification of novel
predictor classifiers for inflammatory bowel disease by gene
expression profiling. PLoS One. 8:e762352013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wright GW and Simon RM: A random variance
model for detection of differential gene expression in small
microarray experiments. Bioinformatics. 19:2448–2455. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Peng J, Wang T, Wang J, Wang Y and Chen J:
Extending gene ontology with gene association networks.
Bioinformatics. 32:1185–1194. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kanehisa M, Sato Y, Kawashima M, Furumichi
M and Tanabe M: KEGG as a reference resource for gene and protein
annotation. Nucleic Acids Res. 44:D457–D462. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Agarwal V, Bell GW, Nam JW and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
ELIFE. 4:2015. View Article : Google Scholar
|
|
12
|
Wong N and Wang X: miRDB: An online
resource for microRNA target prediction and functional annotations.
Nucleic Acids Res. 43(Database issue): D146–D152. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wei ZW, Xia GK, Wu Y, Chen W, Xiang Z,
Schwarz RE, Brekken RA, Awasthi N, He YL and Zhang CH: CXCL1
promotes tumor growth through VEGF pathway activation and is
associated with inferior survival in gastric cancer. Cancer Lett.
359:335–343. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Peter MR, Jerkic M, Sotov V, Douda DN,
Ardelean DS, Ghamami N, Lakschevitz F, Khan MA, Robertson SJ,
Glogauer M, et al: Impaired resolution of inflammation in the
Endoglin heterozygous mouse model of chronic colitis. Mediators
Inflamm. 2014:7671852014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Baugh MD, Perry MJ, Hollander AP, Davies
DR, Cross SS, Lobo AJ, Taylor CJ and Evans GS: Matrix
metalloproteinase levels are elevated in inflammatory bowel
disease. Gastroenterology. 117:814–822. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kusugami K, Nobata K, Tsuzuki T, Ando T
and Ina K: Mucosal expression of matrix metalloproteinases and
their tissue inhibitors in ulcerative colitis patients. J
Gastroenterol. 38:412–413. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
De Bruyn M, Machiels K, Vandooren J,
Lemmens B, Van Lommel L, Breynaert C, van der Goten J, Staelens D,
Billiet T, De Hertogh G, et al: Infliximab restores the
dysfunctional matrix remodeling protein and growth factor gene
expression in patients with inflammatory bowel disease. Inflamm
Bowel Dis. 20:339–352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Biancheri P, Brezski RJ, Di Sabatino A,
Greenplate AR, Soring KL, Corazza GR, Kok KB, Rovedatti L,
Vossenkämper A, Ahmad N, et al: Proteolytic cleavage and loss of
function of biologic agents that neutralize tumor necrosis factor
in the mucosa of patients with inflammatory bowel disease.
Gastroenterology. 149:1564–1574.e3. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Russo I, Luciani A, De Cicco P, Troncone E
and Ciacci C: Butyrate attenuates lipopolysaccharide-induced
inflammation in intestinal cells and Crohn's mucosa through
modulation of antioxidant defense machinery. PLoS One.
7:e328412012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Loddo I and Romano C: Inflammatory bowel
disease: Genetics, epigenetics, and pathogenesis. Front Immunol.
6:5512015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hirano T, Ishihara K and Hibi M: Roles of
STAT3 in mediating the cell growth, differentiation and survival
signals relayed through the IL-6 family of cytokine receptors.
Oncogene. 19:2548–2556. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Aaronson DS and Horvath CM: A road map for
those who don't know JAK-STAT. Science. 296:1653–1655. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Coskun M, Salem M, Pedersen J and Nielsen
OH: Involvement of JAK/STAT signaling in the pathogenesis of
inflammatory bowel disease. Pharmacol Res. 76:1–8. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Warnecke-Eberz U, Chon SH, Hölscher AH,
Drebber U and Bollschweiler E: Exosomal onco-miRs from serum of
patients with adenocarcinoma of the esophagus: Comparison of miRNA
profiles of exosomes and matching tumor. Tumour Biol. 36:4643–4653.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Luo G, Chao YL, Tang B, Li BS, Xiao YF,
Xie R, Wang SM, Wu YY, Dong H, Liu XD and Yang SM: miR-149
represses metastasis of hepatocellular carcinoma by targeting
actin-regulatory proteins PPM1F. Oncotarget. 6:37808–37823.
2015.PubMed/NCBI
|
|
26
|
Wang F, Ma YL, Zhang P, Shen TY, Shi CZ,
Yang YZ, Moyer MP, Zhang HZ, Chen HQ, Liang Y and Qin HL: SP1
mediates the link between methylation of the tumour suppressor
miR-149 and outcome in colorectal cancer. J Pathol. 229:12–24.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang Y, Zheng X, Zhang Z, Zhou J, Zhao G,
Yang J, Xia L, Wang R, Cai X, Hu H, et al: MicroRNA-149 inhibits
proliferation and cell cycle progression through the targeting of
ZBTB2 in human gastric cancer. PLoS One. 7:e416932012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Clay CC, Maniar-Hew K, Gerriets JE, Wang
TT, Postlethwait EM, Evans MJ, Fontaine JH and Miller LA: Early
life ozone exposure results in dysregulated innate immune function
and altered microRNA expression in airway epithelium. PLoS One.
9:e904012014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Carey R, Jurickova I, Ballard E, Bonkowski
E, Han X, Xu H and Denson LA: Activation of an IL-6:STAT3-dependent
transcriptome in pediatric-onset inflammatory bowel disease.
Inflamm Bowel Dis. 14:446–457. 2008. View Article : Google Scholar : PubMed/NCBI
|