Introduction
Gastric cancer (GC) is the fourth most common
malignancy and the third most common cause of cancer-associated
mortality worldwide (1). The
development of GC is a complex and multistep process. It results
from a combination of environmental factors and accumulation of
generalized and specific genetic alterations (2). MicroRNAs (miRNAs or miRs) serve key
roles in post-transcriptional regulation predominantly by pairing
to the 3′-untranslated regions (UTRs) of target mRNA (3). Accumulating evidence suggests that
aberrantly-expressed miRNAs are involved in the initiation,
progression and metastasis of a variety of cancers, including GC
(4–6). Therefore, identification of genes
that regulate tumorigenesis and GC progression are currently under
intense investigation.
The present study investigated miRNAs using
microarray and reverse transcription-quantitative polymerase chain
reaction (RT-qPCR) analyses. Previous work of the authors has
confirmed that miRNA-181a (miR-181a) was significantly upregulated,
whereas myotubularin related protein 3 (MTMR3) was notably
downregulated in human GC tissues and cell lines (7). In addition, ectopic overexpression of
miR-181a promoted tumor progression in SGC-7901 cells (8). Furthermore, MTMR3 was identified as a
direct target of miR-181a. The MTMR3 gene is an essential component
of autophagy (9). However, to
date, the role of MTMR3 in GC remains unclear.
In the current study, aimed to determine the effect
of miR-181a on MTMR3 expression in AGS cells. In addition, the
effect of miR-181a overexpression or MTMR3 knockdown on
proliferation, colony formation, migration and invasion, and
apoptosis of AGS cells was assessed. Furthermore, the effect of
miR-181a and MTMR3 in autophagy was investigated. The results
presented provide evidence that miR-181a and MTMR3 serve important
roles in GC development.
Materials and methods
Cell culture
The AGS human gastric cancer cell line was purchased
from the Cell Bank of Type Culture Collection of Chinese Academy of
Sciences (Shanghai, China). Cells were cultured in RPMI-1640 medium
supplemented with 10% fetal bovine serum, 50 U/ml penicillin and 50
µg/ml streptomycin (Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) at 37°C in humidified 5% CO2 atmosphere.
RNA extraction and RT-qPCR
Total RNA was extracted from AGS cells using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). Reverse
transcription was conducted using the GoScript Reverse
Transcription system (Promega Corporation, Madison, WI, USA).
RT-qPCR was performed using SYBR-Green qPCR SuperMix (Invitrogen;
Thermo Fisher Scientific, Inc.). 18S rRNA was used as an internal
control. PCR primers used were as follows: MTMR3, forward
5′-GCAGGACCAGATATGTGAGAGA-3′ and reverse
5′-GCACACAACATGCAGATGAG-3′; 18S rRNA, forward
5′-CCTGGATACCGCAGCTAGGA-3′ and reverse 5′-GCGGCGCAATACGAATGCCCC-3′.
RT-qPCR was performed at 95°C for 10 min, followed by 40 cycles of
95°C for 15 sec and 60°C for 45 sec. The RT-qPCR kit and protocol
for miR-181a have been described in detail previously (7). RT-qPCR was detected using the ABI
PRISM 7500 Sequence Detection system (Applied Biosystems; Thermo
Fisher Scientific, Inc.). Each experiment was repeated three times
in duplicate. The relative quantification was calculated using the
2-∆∆Ct method (10).
Oligonucleotide construction and
transfection
All RNA oligonucleotides were synthesized and
purchased from Shanghai GenePharma Co., Ltd. (Shanghai, China).
Oligonucleotides sequences used were as follows: miR-181a mimic,
AACAUUCAACGCUGUCGGUGAGU; miR-181a inhibitor,
ACCAUCGACCGUUGAUUGUACC; negative controls (NC),
UUCUCCGAACGUGUCACGUTT and ACGUGACACGUUCGGAGAATT; NC inhibitor,
CAGUACUUUUGUGUAGUACAA; MTMR3 siRNA (siMTMR3),
GGAAGAUAAGGUGAAGUCAdTdT and UGACUUCACCUUAUCUUCCdTdT; scramble
mimic, UUCUCCGAACGUGUCACGUTT and ACGUGACACGUUCGGAGAATT. Cells were
transfected with miR-181a mimic (50 nM), miR-181a inhibitor (100
nM), miRNA NC (50 nM), NC inhibitor (100 nM), siMTMR3 (400 nM) or
scramble mimic (400 nM) using Lipofectamine™ RNAiMAX (Invitrogen;
Thermo Fisher Scientific, Inc.). Following 5 h incubation, the
medium in each well was replaced by serum-containing medium. Total
RNA and protein was extracted 48 h following transfection.
Western blot analysis
Total protein was extracted using Cell Lysis
reagents (Pierce; Thermo Fisher Scientific, Inc.) and quantified by
the bicinchoninic acid method (Pierce; Thermo Fisher Scientific,
Inc.). Proteins were separated on 10% SDS-polyacrylamide gel, and
then transferred to a polyvinylidene difluoride membrane (GE
Healthcare Life Sciences, Chalfont, UK). The membrane was blocked
in 5% non-fat dry milk in TBS and Tween-20 for 1 h at room
temperature and incubated with anti-MTMR3 antibody (dilution,
1:400; Novus Biologicals, LLC, Littleton, CO, USA; cat. no.
H00008897-B01P), anti-microtubule associated protein 1 light chain
3 beta (LC3B) antibody (cat. no. L7543; dilution, 1:500;
Sigma-Aldrich; Merck Millipore, Darmstadt, Germany) or anti-GAPDH
antibody (; cat. no. ab9485; dilution, 1:2,500; Abcam, Cambridge,
UK) overnight at 4°C, followed by incubation with rabbit anti-mouse
IgG and swine anti-rabbit IgG secondary antibodies, both conjugated
with horseradish peroxidase (Dako; Agilent Technologies, Inc.,
Santa Clara, CA, USA; cat. nos. F00037626 and A00048749) for 50 min
at room temperature. The membranes were developed using the ECL kit
(Pierce; Thermo Fisher Scientific, Inc.) and exposed to X-ray film
to visualize the images. The GAPDH gene was used as an internal
control. The band intensity was analyzed using Gel-Pro Analyzer
software (version 4.0; Media Cybernetics, Inc., Rockville, MD,
USA).
Proliferation assay
At 24 h following transfection, cells were
trypsinized and seeded into 96-well plates at a density of
1×104 cells/well in growth medium supplemented with 10%
serum. The cell proliferation was determined at different time
points (24, 48, 72 and 96 h) using the MTS kit (CellTiter 96
Aqueous One Cell Proliferation Assay; Promega Corporation)
following the manufacturer's instructions, and the absorption was
read at 490 nm.
Colony formation assay
At 24 h after transfection, cells were trypsinized
and seeded onto 96-well plates at a density of 200 cells/well. The
cells were observed every 24 h. Following 7 days of incubation, the
colonies were fixed with 4% paraformaldehyde and then stained with
crystal violet for 15 min. The ELISPOT reader (iSPOT system;
Autoimmun Diagnostika GmbH, Strassberg, Germany) was used to read
the plates. The colony formation rate was acquired through number
of colonies/number of plated cells.
Cell cycle assay
Cells were harvested by trypsinization 48 h after
transfection, washed three times with ice-cold PBS, and fixed with
70% ethanol overnight at 4°C. The fixed cells were washed in PBS
and subjected to propidium iodide (PI)/RNase A staining followed by
flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA). The
percentage of cells in each phase of the cell cycle was estimated
using Modfit LT software, (version, 3.0; BD Biosciences).
Apoptosis assay
Following 24 h post-transfection, cells were labeled
with Annexin V-fluorescein isothiocyanate and PI (Nanjing KeyGen
Biotech. Co., Ltd., Nanjing, China), according to the
manufacturer's instructions. Cells were analyzed by flow cytometry
(BD Biosciences).
Cell migration and invasion assay
Cell migration assay was performed using 8 µm-pore
size 96-well MIC Transwell plates (EMD Millipore, Billerica, MA,
USA). Following transfection, the cells were resuspended in
serum-free medium and 1×105 cells in 100 µl medium were
added to the upper chamber while the lower chamber was filled with
complete media as a chemoattractant. The cells were incubated for
24 h at 37°C, the cells on the upper surface of the membrane were
removed by cotton swabs and the cells attached to the lower surface
were fixed in ice-cold methanol for 10 min, and stained with 0.5%
crystal violet solution for 10 min. Then the number of migrated
cells on the lower surface of the membrane was counted under a
microscope in five fields. For the invasion assay, the MIC plates
were initially coated with Matrigel (BD Biosciences) diluted in
serum free medium and the same procedures as migration assay were
performed.
GFP-LC3 analysis
Cells were transfected with a pSELECT-GFP-LC3
expression vector plasmid (InvivoGen, San Diego, CA, USA) using
Lipofectamine reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
following the manufacturer's instructions. In the starvation
experiment, 24 h after co-transfection of miRNAs and GFP-LC3, cells
were incubated for 2 h in Earle's Balanced Salt solution medium.
Cells were fixed with 4% paraformaldehyde in PBS after the
treatments. Images were obtained using a fluorescent microscope.
Cells with more than five intense GFP-LC3 puncta were considered
autophagic, as LC3 is a marker for autophagosomes, whereas those
with diffuse cytoplasmic GFP-LC3 staining were considered
non-autophagic. The percentage of GFP-LC3-positive cells were
counted in >100 cells.
Statistical analysis
Differences between two groups were analyzed by
Student's t-test. Data were represented as the mean ± standard
deviation from three independent experiments. Statistical tests
were two-tailed. P<0.05 was considered to indicate a
statistically significant difference, and was analyzed using SPSS
software (version, 17.0; SPSS, Inc., Chicago, IL, USA).
Results
miR-181a inhibits MTMR3 expression in
AGS cells
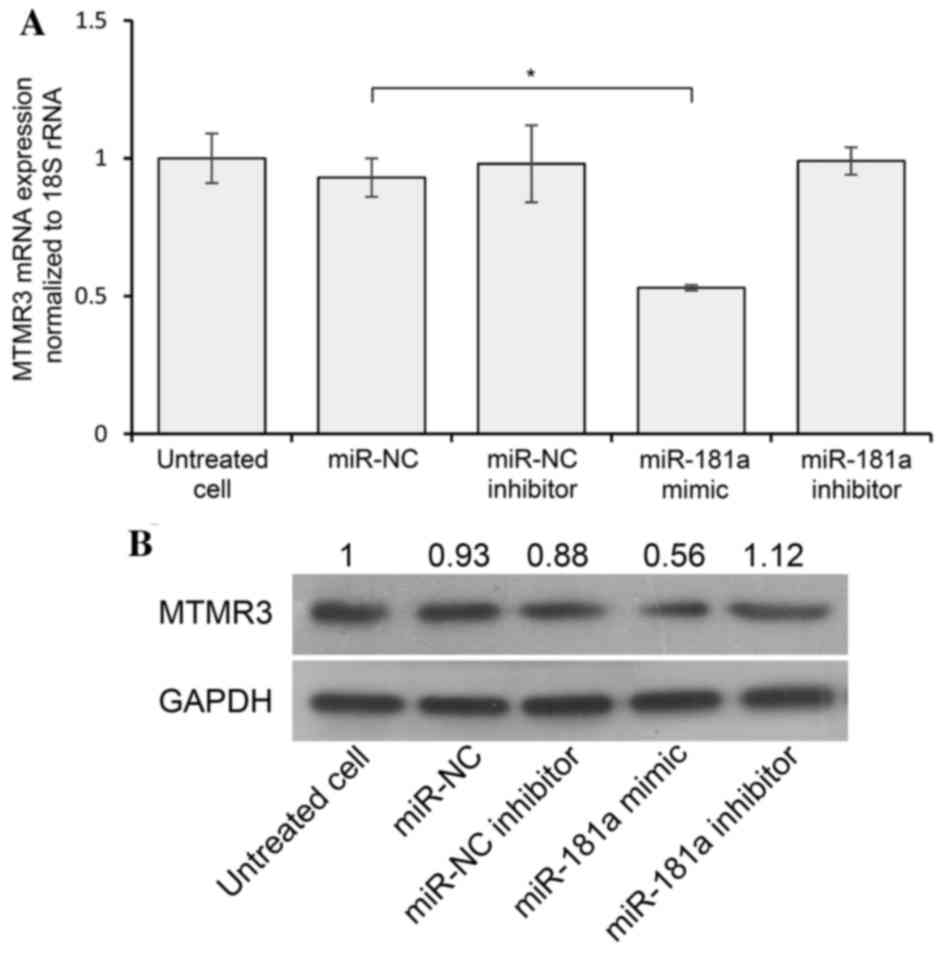
A previous study demonstrated that miR-181a directly
targeted the 3′-UTR of MTMR3 and suppressed the expression of MTMR3
by dual-luciferase assays in HEK-293T cells. In addition, the study
validated that artificial overexpression of miR-181a effectively
reduced the levels of MTMR3 protein in SGC-7901 cells (7). To further confirm the interaction
between miR-181a and MTMR3 in AGS cells, RT-qPCR and western blot
analysis were performed, which demonstrated that transfection of
miR-181a led to a decrease in MTMR3 mRNA and protein levels
compared with the negative control (Fig. 1A and B). However, inhibition of
endogenous miR-181a did not increase the levels of MTMR3 mRNA and
protein, suggesting that MTMR3 may be suppressed via other factors
in addition to miR-181a. These data suggest that MTMR3 is a target
of miR-181a.
Effect of overexpressed miR-181a or
silenced MTMR3 on proliferation, colony formation, cell cycle,
apoptosis, migration and invasion of AGS cells
To explore the role of miR-181a and MTMR3 on the
growth of GC cells, an miR-181a mimic or siMTMR3 was transfected
into AGS cells. The results of the MTS assay indicated that the
proliferation capacity of AGS cells was promoted by upregulation of
miR-181a or downregulation of MTMR3, compared with the respective
controls (Fig. 2A). Colony
formation assay suggested that the number of colonies was
significantly higher for cells transfected with the miR-181a mimic
and siMTMR3 compared with their respective controls (P<0.05;
Fig. 2B and C). To assess whether
this effect is mediated through perturbation of the cell cycle,
cell cycle distribution analysis was performed. There was no
difference identified in cell cycle profile among the four groups
(miR-NC, miR-181a, scramble and siMTMR3; Fig. 2D). Furthermore, flow cytometry
analysis was used to detect the apoptosis of AGS cells transfected
with miR-181a mimic or siMTMR3. It demonstrated that the apoptosis
rates were decreased in the miR-181a and siMTMR3 groups compared
with the NC and scramble groups, respectively (P<0.05; Fig. 2E and F). Finally, results of the
Transwell migration assay revealed that artificial overexpression
of miR-181a or reduction of MTMR3 significantly increased the
migration potential of AGS cells (Fig.
2G). In addition, a significantly higher number of cells in
miR-181a and siMTMR3 groups were found to cross the Matrigel
compared with the controls in the invasion assay (Fig. 2H). These results indicate that
MTMR3 knockdown phenocopied the effect of miR-181a overexpression
in promoting the development of AGS.
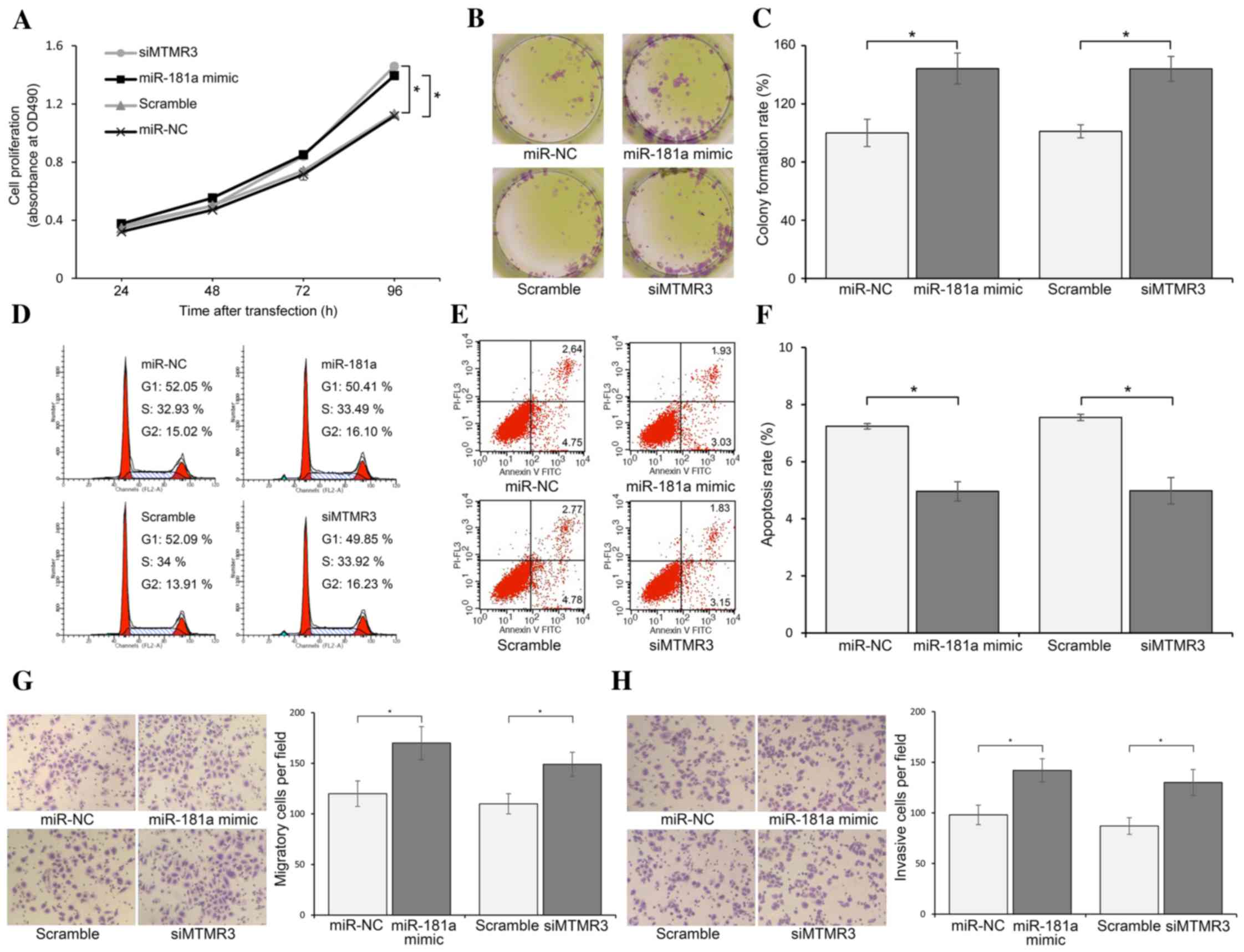 | Figure 2.Overexpressed miR-181a or silenced
MTMR3 promote proliferation, colony formation, migration and
invasion, and inhibit apoptosis of AGS cells. (A) MTS assay showing
the proliferation of AGS cells when treated as indicated, measured
at 24, 48, 72 and 96 h following transfection. (B) Colony formation
of AGS cells treated as indicated. The colonies were counted on day
7 following transfection. (C) The colony formation rate was
acquired through number of colonies/number of planted cells. (D)
Cell cycle analysis by flow cytometry. There was no difference in
cell cycle distribution among four groups. (E) Flow cytometric
assay for the evaluation of apoptosis of AGS cells treated as
indicated. Annexin V-FITC+/PI− population
(lower right quadrant in diagram) indicated early stage apoptosis,
and Annexin V-FITC+/PI+ population (upper
right quadrant in diagram) indicated late stage apoptosis. (F)
Total apoptosis rate was equal to early stage apoptosis + late
stage apoptosis. (G) Migration and (H) invasion assay of AGS cells
treated as indicated. After 24 h, the number of cells that had
migrated/invaded through the membrane were counted under a
microscope using five random fields (magnification, ×100). The
results represent the means ± standard deviation for three
independent experiments. *P<0.05 miR-181a mimic vs. miR-NC, and
siMTMR3 vs. Scramble. siMTMR3, myotubularin related protein 3 small
interfering RNA; miR, microRNA; NC, negative control; FITC,
fluorescein isothiocyanate; PI, propidium iodide. |
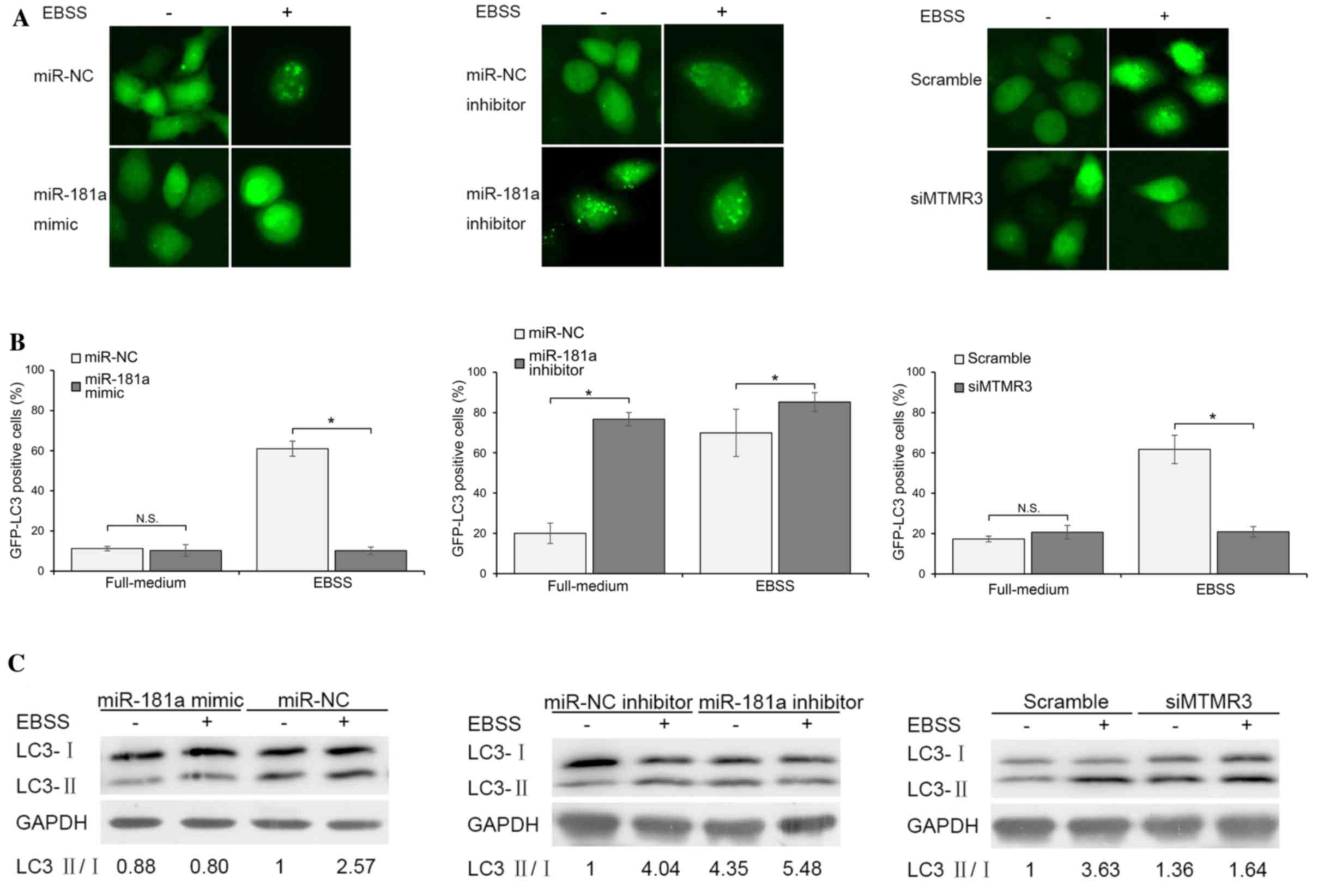
miR-181a and MTMR3 regulate autophagy
in AGS cells
Autophagy was determined by following the
redistribution of GFP-LC3 from a diffuse to a punctate pattern or
by assessing the conversion of endogenous LC3 protein from the
cytosolic LC3-I to the autophagosome associated LC3-II by
immunoblotting. As presented in Fig.
3A and B, following co-transfection of the GFP-LC3 plasmid and
miR-NC/miR-181a mimic, GFP-LC3 was observed predominantly as
diffuse green fluorescence in the cytoplasm, indicating low
autophagic activities in both groups. This phenomenon was confirmed
by immunoblotting (Fig. 3C). Cells
were incubated in EBSS medium for 2 h to induce autophagy. As
presented in Fig. 3A and B,
overexpression of miR-181a significantly blocked starvation-induced
GFP-LC3 dot accumulation. In line with this result,
starvation-activated lipidation of LC3-II was attenuated in
miR-181a mimic transfection group (Fig. 3C). To further demonstrate the
effects of endogenous miR-181a inhibition on autophagy, cells were
transfected with miR-181a inhibitor or miR-NC inhibitor and
autophagy was observed under non-starved or starved conditions.
Results indicated that cells treated with the miR-181a inhibitor
exhibited a significantly higher percentage of punctate GFP and
LC3-II conversion than cells of miR-NC inhibitor group. In
addition, the inhibition of miR-181a further increased autophagic
activities during starvation compared with NC (Fig. 3A and B). The role of miR-181a
target gene MTMR3 in autophagy was then evaluated. The effect of
silenced MTMR3 was similar to that of overexpressed miR-181a,
although to a lesser extent (Fig.
3A-C). Knockdown of MTMR3 significantly suppressed
starvation-induced autophagy. Together, these findings suggested
that miR-181a is a negative regulator of autophagy, partially
mediated by inhibition of MTMR3.
Discussion
Various previous studies have demonstrated that the
aberrant expression of miRNAs is implicated in human malignancies
(2,11,12).
Identification of cancer-specific miRNAs and their targets is
critical for understanding tumorigenesis, and may be important for
defining novel therapeutic targets. miR-181a, a member of the
miR-181 family, is involved in various events, including
development, differentiation, hematopoiesis and immune modulation
(13,14). The diverse functions of miR-181 in
human carcinogenesis may be due to different target genes,
depending on the tissue or cellular environment. miR-181a is
downregulated in lung cancer (15), leukemia (16) and glioblastoma (17) and acts as a tumor suppressor by
targeting KRAS (18), Bcl-2
(19) and PLAG1 (16). In addition, miR-181a is identified
as an oncogene by targeting ATM serine/threonine kinase (20), CDX2 (13) and KLF6 (8), and is upregulated in pancreatic
cancer (21), hepatocellular
carcinoma (13) and gastric cancer
(22). The present study revealed
that miR-181a functioned as an oncogene in GC cells. Overexpression
of miR-181a enhanced the capacity of AGS cells for proliferation,
colony formation, migration and invasion, whereas it attenuated
apoptosis and autophagy. Similarly, Chen et al (22) indicated that enforced expression of
miR-181a promoted GC cells proliferation ability. Tekirdag et
al (14) suggested that
overexpression of miR-181a results in the attenuation of
starvation- and rapamycin-induced autophagy in MCF-7, Huh-7 and
K562 cells. Furthermore, inhibition of endogenous miR-181a
stimulated autophagy. Given that a single miRNA has many different
targets, the authors hypothesize that miR-181a targets multiple
genes during the regulation of the malignant biological behavior of
GC cells, and MTMR3 is one of these targeted genes.
MTMR3 is a member of myotubularin-related protein
family, with at least 11 phosphatidylinositol 3-phosphate (PI3P)
phosphatases proteins in human. The synthesis of PI3P is required
for the initiation of autophagy (23). Autophagy is a cellular degradation
pathway for the clearance of damaged or superfluous proteins and
organelles. The role of autophagy in cancer is a double-edged sword
(24). Certain reports indicate
that autophagy serves as a cell survival mechanism, whilst others
suggested that autophagy induces autophagic cell death (25,26).
A recent study demonstrated that MTMR3 decreased pattern
recognition receptor (PRR)-induced PI3P and autophagy levels in
monocyte-derived macrophages. In MTMR3-deficient macrophages,
reducing the enhanced autophagy or restoring nuclear factor-κB
signaling rescued PRR-induced cytokines. Thus, modulation of MTMR3
levels may provide a therapeutic benefit in inflammatory bowel
disease (27). Another study
demonstrated that MTMR3 expression was significantly reduced in
colonic biopsies from ulcerative colitis patients (28). Because autophagy may be a tumor
suppressor or promoter, the exact function of MTMR3 in cancer
remains elusive. A report highlighted that microsatellite
instability in MTMR3 may serve a role in the development of gastric
and colorectal cancer (29).
Another study indicated that MTMR3 was a miR-99a target, and
reduced MTMR3 expression in the oral cancer line significantly
attenuated cell proliferation, migration, and invasion (26). In addition, miR-100 was
demonstrated to increase proliferation and decrease apoptosis of
breast cancer cells via targeting MTMR3 (30). The data presented in the current
study demonstrated that silencing MTMR3 led to promotion of
proliferation, colony formation, migration and invasion, and
inhibition of apoptosis and autophagy of GC cells. Further studies
are required to clarify the in vivo role of MTMR3 in GC.
In summary, the current work provides evidence that
miR-181a and MTMR3 are important during tumorigenesis and
progression of AGS cells. In addition, miR-181a was identified as a
novel autophagy regulating miRNA and may act by targeting MTMR3.
The results may help understand the potential molecular mechanisms
of gastric cancer development and may have potential therapeutic
value in the future.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81302078).
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wan X, Ding X, Chen S, Song H, Jiang H,
Fang Y, Li P and Guo J: The functional sites of miRNAs and lncRNAs
in gastric carcinogenesis. Tumour Biol. 36:521–532. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brodersen P and Voinnet O: Revisiting the
principles of microRNA target recognition and mode of action. Nat
Rev Mol Cell Biol. 10:141–148. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shen J, Xiao Z, Wu WK, Wang MH, To KF,
Chen Y, Yang W, Li MS, Shin VY, Tong JH, et al: Epigenetic
silencing of miR-490-3p reactivates the chromatin remodeler SMARCD1
to promote Helicobacter pylori-induced gastric carcinogenesis.
Cancer Res. 75:754–765. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yu Z, Zhang W and Deng F: MicroRNA-577
inhibits gastric cancer growth by targeting E2F transcription
factor 3. Oncol Lett. 10:1447–1452. 2015.PubMed/NCBI
|
|
6
|
Song H, Xu W, Song J, Liang Y, Fu W, Zhu
XC, Li C, Peng JS and Zheng JN: Overexpression of Lin28 inhibits
the proliferation, migration and cell cycle progression and induces
apoptosis of BGC-823 gastric cancer cells. Oncol Rep. 33:997–1003.
2015.PubMed/NCBI
|
|
7
|
Lin Y, Nie Y, Zhao J, Chen X, Ye M and Li
Y, Du Y, Cao J, Shen B and Li Y: Genetic polymorphism at miR-181a
binding site contributes to gastric cancer susceptibility.
Carcinogenesis. 33:2377–2383. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang X, Nie Y, Du Y, Cao J, Shen B and Li
Y: MicroRNA-181a promotes gastric cancer by negatively regulating
tumor suppressor KLF6. Tumour Biol. 33:1589–1597. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Henderson P, van Limbergen JE, Wilson DC,
Satsangi J and Russell RK: Genetics of childhood-onset inflammatory
bowel disease. Inflamm Bowel Dis. 17:346–361. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ueda T, Volinia S, Okumura H, Shimizu M,
Taccioli C, Rossi S, Alder H, Liu CG, Oue N, Yasui W, et al:
Relation between microRNA expression and progression and prognosis
of gastric cancer: A microRNA expression analysis. Lancet Oncol.
11:136–146. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li Y, Kuscu C, Banach A, Zhang Q,
Pulkoski-Gross A, Kim D, Liu J, Roth E, Li E, Shroyer KR, et al:
miR-181a-5p inhibits cancer cell migration and angiogenesis via
downregulation of matrix metalloproteinase-14. Cancer Res.
75:2674–2685. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ji J, Yamashita T, Budhu A, Forgues M, Jia
HL, Li C, Deng C, Wauthier E, Reid LM, Ye QH, et al: Identification
of microRNA-181 by genome-wide screening as a critical player in
EpCAM-positive hepatic cancer stem cells. Hepatology. 50:472–480.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tekirdag KA, Korkmaz G, Ozturk DG, Agami R
and Gozuacik D: MIR181A regulates starvation- and rapamycin-induced
autophagy through targeting of ATG5. Autophagy. 9:374–385. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gao W, Shen H, Liu L, Xu J, Xu J and Shu
Y: MiR-21 overexpression in human primary squamous cell lung
carcinoma is associated with poor patient prognosis. J Cancer Res
Clin Oncol. 137:557–566. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pallasch CP, Patz M, Park YJ, Hagist S,
Eggle D, Claus R, Debey-Pascher S, Schulz A, Frenzel LP, Claasen J,
et al: miRNA deregulation by epigenetic silencing disrupts
suppression of the oncogene PLAG1 in chronic lymphocytic leukemia.
Blood. 114:3255–3264. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ciafrè SA, Galardi S, Mangiola A, Ferracin
M, Liu CG, Sabatino G, Negrini M, Maira G, Croce CM and Farace MG:
Extensive modulation of a set of microRNAs in primary glioblastoma.
Biochem Biophys Res Commun. 334:1351–1358. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shin KH, Bae SD, Hong HS, Kim RH, Kang MK
and Park NH: miR-181a shows tumor suppressive effect against oral
squamous cell carcinoma cells by downregulating K-ras. Biochem
Biophys Res Commun. 404:896–902. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhu W, Shan X, Wang T, Shu Y and Liu P:
miR-181b modulates multidrug resistance by targeting BCL2 in human
cancer cell lines. Int J Cancer. 127:2520–2529. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Y, Yu Y, Tsuyada A, Ren X, Wu X,
Stubblefield K, Rankin-Gee EK and Wang SE: Transforming growth
factor-β regulates the sphere-initiating stem cell-like feature in
breast cancer through miRNA-181 and ATM. Oncogene. 30:1470–1480.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang P, Guo Z, Hu R, He X, Jiao X and Zhu
X: Interaction between microRNA-181a and TNFAIP1 regulates
pancreatic cancer proliferation and migration. Tumour Biol. 2015.
View Article : Google Scholar
|
|
22
|
Chen G, Shen ZL, Wang L, Lv CY, Huang XE
and Zhou RP: Hsa-miR-181a-5p expression and effects on cell
proliferation in gastric cancer. Asian Pac J Cancer Prev.
14:3871–3875. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vergne I and Deretic V: The role of PI3P
phosphatases in the regulation of autophagy. FEBS Lett.
584:1313–1318. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
White E: Deconvoluting the
context-dependent role for autophagy in cancer. Nat Rev Cancer.
12:401–410. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kuo YZ, Tai YH, Lo HI, Chen YL, Cheng HC,
Fang WY, Lin SH, Yang CL, Tsai ST and Wu LW: MiR-99a exerts
anti-metastasis through inhibiting myotubularin-related protein 3
expression in oral cancer. Oral Dis. 20:e65–e75. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lahiri A, Hedl M and Abraham C: MTMR3 risk
allele enhances innate receptor-induced signaling and cytokines by
decreasing autophagy and increasing caspase-1 activation. Proc Natl
Acad Sci USA. 112:10461–10466. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Imielinski M, Baldassano RN, Griffiths A,
Russell RK, Annese V, Dubinsky M, Kugathasan S, Bradfield JP,
Walters TD, Sleiman P, et al: Common variants at five new loci
associated with early-onset inflammatory bowel disease. Nat Genet.
41:1335–1340. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Song SY, Kang MR, Yoo NJ and Lee SH:
Mutational analysis of mononucleotide repeats in dual specificity
tyrosine phosphatase genes in gastric and colon carcinomas with
microsatellite instability. APMIS. 118:389–393. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gong Y, He T, Yang L, Yang G, Chen Y and
Zhang X: The role of miR-100 in regulating apoptosis of breast
cancer cells. Sci Rep. 5:116502015. View Article : Google Scholar : PubMed/NCBI
|