Introduction
Primary hypertrophic osteoarthropathy (PHO; MIM
259100 and 614441), also known as pachydermoperiostosis, is a rare
multi-organic, disease primarily characterized by digital clubbing,
periostosis and pachydermia (1).
The manifestations usually begin at puberty and progress gradually
over years prior to stabilization of the disease. As the primary
form of hypertrophic osteoarthropathy, PHO is distinct from the
more common secondary hypertrophic osteoarthropathy, which is
always associated with an underlying cause, for example pulmonary
or cardiac disease. In terms of the inheritance of PHO, autosomal
dominant with incomplete penetrance and recessive inheritance have
previously been confirmed (2,3).
However, the precise incidence and prevalence remain to be
elucidated (4).
PHO is a genetically heterogeneous disease. To date,
two genes have been reported to be associated with PHO (5,6). In
2008, 15-hydroxyprostaglandin dehydrogenase (HPGD; MIM
601688), which encodes 15-PGDH, was identified as the primary
causative factor of PHO. Zhang et al (6) demonstrated another pathogenic gene
responsible for PHO, solute carrier organic anion transporter
family, member 2A1 (SLCO2A1; MIM 601460), which is a gene
encoding the prostaglandin transporter. It has been suggested that
the two genes, HPGD and SLCO2A1, are involved in the
degradation of prostaglandin E2 (PGE2). The degradation of PGE2
involves two steps (7): Selective
uptake across the plasma membrane by SLCO2A1 and degradation
inside the cell by HPGD. Considering these molecular
findings, PHO has been categorized into two types (1): i) hypertrophic osteoarthropathy,
primary, autosomal recessive, type 1 (PHOAR1; MIM 259100) due to
HPGD deficiency; and ii) hypertrophic osteoarthropathy,
primary, autosomal recessive, type 2 (PHOAR2; MIM 614441) due to
SLCO2A1 deficiency.
When examining a patient with digital clubbing, it
is important to differentiate between secondary hypertrophic
osteoarthropathy due to pulmonary disease or other conditions,
which is more common, and PHO, which is less common. However,
several clinical examinations are required to eliminate secondary
causes, which is an expensive and time-consuming process. In
addition, as PHO is clinically rare and the disease remains to be
fully understood, it can lead to a missed diagnosis or misdiagnosis
and consequent suboptimal treatment. At present, the identification
of SLCO2A1 or HPGD deficiency in patients with PHO
has become an efficient approach to assist in clinical diagnoses.
However, the genotype/phenotype associations have not been
observed, and the clinical phenotype and mutational spectrum of
these pathogenic genes require further investigation in order to
fully understand this disease. The present study reported on a
20-year-old Chinese patient with PHO. The study identified two
novel mutations (c.349delC/p.L117SfsX56 and c.1286A>G/p.Y429C)
in the SLCO2A1 gene in PHO disease. To the best of our
knowledge, these mutations have not been reported in a previous
study, or presented in the Single Nucleotide Polymorphism Database
(dbSNP) (http://www.ncbi.nlm.nih.gov/projects/SNP) or the Human
Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php).
Materials and methods
Clinical findings
The protocol for the present study was approved by
the Review Board of The Second Xiangya Hospital of the Central
South University (Changsha, China), and the participants involved
provided informed written consent. All experiments were performed
in accordance with relevant guidelines and regulations. A
20-year-old man was born to healthy non-consanguineous parents.
Apparently healthy during his childhood, he reported that at 18
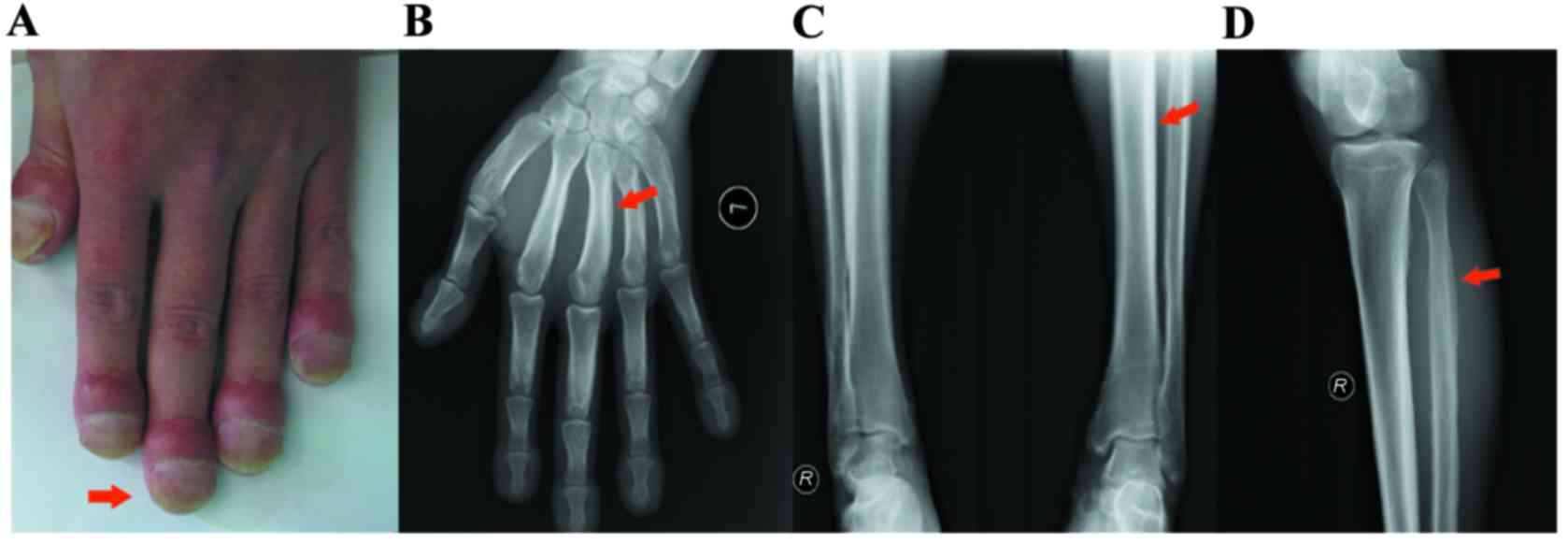
years of age, he noticed progressive enlargement of the terminal
phalanges and pachydermia (Fig.
1A). After 2 years, he gradually presented with joint
discomfort and action inconvenience. He denied any gastrointestinal
discomfort, blepharitis, seborrhea, or hyperhidrosis. Radiography
revealed periosteal overgrowth of the tibiofibular joint and a
region of the metacarpus (Fig.
1B-D). Physical examination revealed no other secondary
hypertrophic osteoarthropathy, including congenital heart disease
or lung abnormities. All other laboratory investigations were
within normal limits, including complete blood count, liver
function test, renal profile, thyroid stimulating hormone and
growth hormone. The patient had no siblings, and his parents were
healthy without the features described above.
Sequence analysis of associated
genes
Informed consent was obtained from the patient and
from 200 healthy volunteers (19–54 years old; 108 male, 92 female;
recruited from the physical examination center of the Second
Xiangya Hospital of Central South Hospital, Changsha, China) prior
to blood sampling and DNA analysis. The DNA was extracted from
peripheral white blood cells using conventional methods. The DNA
sequences of SLCO2A1 and HPGD were obtained from the
Ensembl online database (http://asia.ensembl.org/index.html; Ensembl nos.
ENSG00000174640 and ENSG00000164120, respectively). Primers for
polymerase chain reaction (PCR) were designed using IDT software
(http://www.idtdna.com/site) and the
sequences are listed in Table I.
The entire coding regions, including the flanking intronic
sequences of SLCO2A1 and HPGD, were amplified by PCR.
Patient DNA samples were amplified in 25 µl reactions using 2X
Power Taq PCR MasterMix (12.5 µl; Bioteke Corporation, Beijing,
China), nuclease-free water (11 µl), 10 pmol/ul forward and reverse
primers (0.5 µl each), and 100 ng/µl template (0.5 µl).
Thermocycling conditions were as follows: Initial denaturation at
94°C for 5 min, 35 cycles of denaturation at 94°C (30 sec),
annealing temperature (30 sec) (Table
I), extension at 72°C (1 min), and final extension at 72°C (7
min). Sequences of the PCR products were determined using the ABI
3100 genetic analyzer (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) as previously described (8).
To assess the damaging effects of variants in silico, the
online databases, Polyphen2 (http://genetics.bwh.harvard.edu/pph2) (9) and MutationTaster (www.mutationtaster.org) (10), were used to predict the possible
effects of variants.
 | Table I.Primer sequences for polymerase chain
reaction. |
Table I.
Primer sequences for polymerase chain
reaction.
| Gene | Primer sequence
(5′→3′) | Annealing temperature
(°C) |
|---|
| SLCO2A1 (1) | F:
ACCCTCATATCCCAGCCTTC | 59.8 |
|
| R:
TGTCCGAGTAAGCGGTAAGC | 59.8 |
| SLCO2A1 (2) | F:
CACTGGGCCACATATCACAG | 59.8 |
|
| R:
CTGTTACCCGGCAGAAAGAG | 59.8 |
| SLCO2A1 (3) | F:
GGAGATGGAGACCCAGAAGG | 61.9 |
|
| R:
GCACACTTTCCTGAACAAACC | 58.0 |
| SLCO2A1 (4) | F:
CAGGAACCATGTCCCATTTG | 57.8 |
|
| R:
ACACAGCTGGGAGGTAATGG | 59.8 |
| SLCO2A1 (5) | F:
ACAGGTGTGGGCTTATCAGG | 59.8 |
|
| R:
CAGCAGCTTGTTCCTCACAG | 59.8 |
| SLCO2A1 (6) | F:
CCTCTGGGAAGACCAATAGC | 59.8 |
|
| R:
TGGAGGTCTCCTGATCCTTG | 59.8 |
| SLCO2A1 (7) | F:
GGAAATGCAGGTGCTGTTTG | 57.8 |
|
| R:
TCTGCTCCTACTGTCCCTTAC | 60.0 |
| SLCO2A1 (8) | F:
CCCTGTGGTGTTGTGTGC | 59.6 |
|
| R:
CTGACTGGAAGGACAGGAG | 59.7 |
| SLCO2A1 (9) | F:
GCCTGGCAAGCAGTAAATG | 57.6 |
|
| R:
TGCTTGAACCTGGGAGAATC | 57.8 |
| SLCO2A1 (10) | F:
AAATGGAGAGATGCCGTGAC | 57.8 |
|
| R:
CCCAGGGTAGGGAGGTAGAG | 64.0 |
| SLCO2A1 (11) | F:
TTGCCCAAACAGTGACAGAG | 57.8 |
|
| R:
CCTGCAATGAGGAGCTCAG | 59.7 |
| SLCO2A1 (12) | F:
TAGAGCATTCAGCCCAGGTG | 59.8 |
|
| R:
CCTCAAGCAATCTGGGAAAC | 57.8 |
| SLCO2A1 (13) | F:
GCCCGTGTATCTCCACTCTG | 61.9 |
|
| R:
TGGCCCTTCATGTTCTCTTC | 57.8 |
| SLCO2A1 (14) | F:
CCTGCTTCCCTACAGCTTTG | 59.8 |
|
| R:
GGGTACACAGTGGCCCTTAG | 61.9 |
| HPGD (1) | F:
GCTGGCTTGACAGTTTCCTC | 59.8 |
|
| R:
AGTCTCGGAGTGTGTGGGC | 61.9 |
| HPGD (2) | F:
GTGTTTATTGTTTGTCCGTCTA | 54.5 |
|
| R:
CAGTCTTGCCTTTCTTTCG | 55.4 |
| HPGD (3) | F:
CCTCTCATGGCATAGGACATG | 60.0 |
|
| R:
GTTTCCATGACTCCAAGAACC | 58.0 |
| HPGD (4) | F:
CCACAATGATTAGGCAAAC | 53.2 |
|
| R:
AAGCCACAAGTTAAATTAAGAG | 52.6 |
| HPGD (5) | F:
AAATTCTGGACGACACGG | 55.0 |
|
| R:
TTCCACCTTTCATCCAAGT | 53.2 |
| HPGD (6) | F:
TTGTTACATAGCTGGGAG | 52.7 |
|
| R:
ATAATGCTTTGCTTCATC | 48.2 |
| HPGD (7) | F:
ATGCCTCATTCTTTCGTT | 50.5 |
|
| R:
TAGCCTTTGGTCCACATC | 55.0 |
Results
In the present study the SLCO2A1 and
HPGD mutations were screened for in the patient and his
parents using PCR followed by direct sequence analysis (Fig. 2). Sequencing revealed compound
heterozygous mutations in the SLCO2A1 gene, which consisted
of a novel frameshift deletion, p.L117SfsX56 (c.349 delC), in exon
3 (Fig. 2A) and a novel missense
mutation, p.Y429C (c.1286A>G), in exon 9 (Fig. 2B). The p.L117SfsX56 mutation
resulted in a frameshift at amino acid position 117 and the
introduction of a premature stop codon after 56 amino acid
residues, and was predicted to be disease-causing by MutationTaster
(www.mutationtaster.org), with a
probability value of 1. The p.Y429C mutation was identified as
non-conservative, affected evolutionarily conserved amino acids in
diverse species (Fig. 2I) and was
predicted in silico by all bioinformatics tools used to be
of pathogenic relevance (PolyPhen-2 score, 0.999; MutationTaster
score, 0.999). Based on the DNA sequence analysis, the p.L117SfsX56
and p.Y429C mutations were found to be inherited from the patient's
unaffected father and mother, respectively, and were not found in
the cohort of 200 control individuals (Fig. 2C-H) (8). Neither of these mutations has been
reported in a previous study, or was present in the dbSNP or Human
Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php).
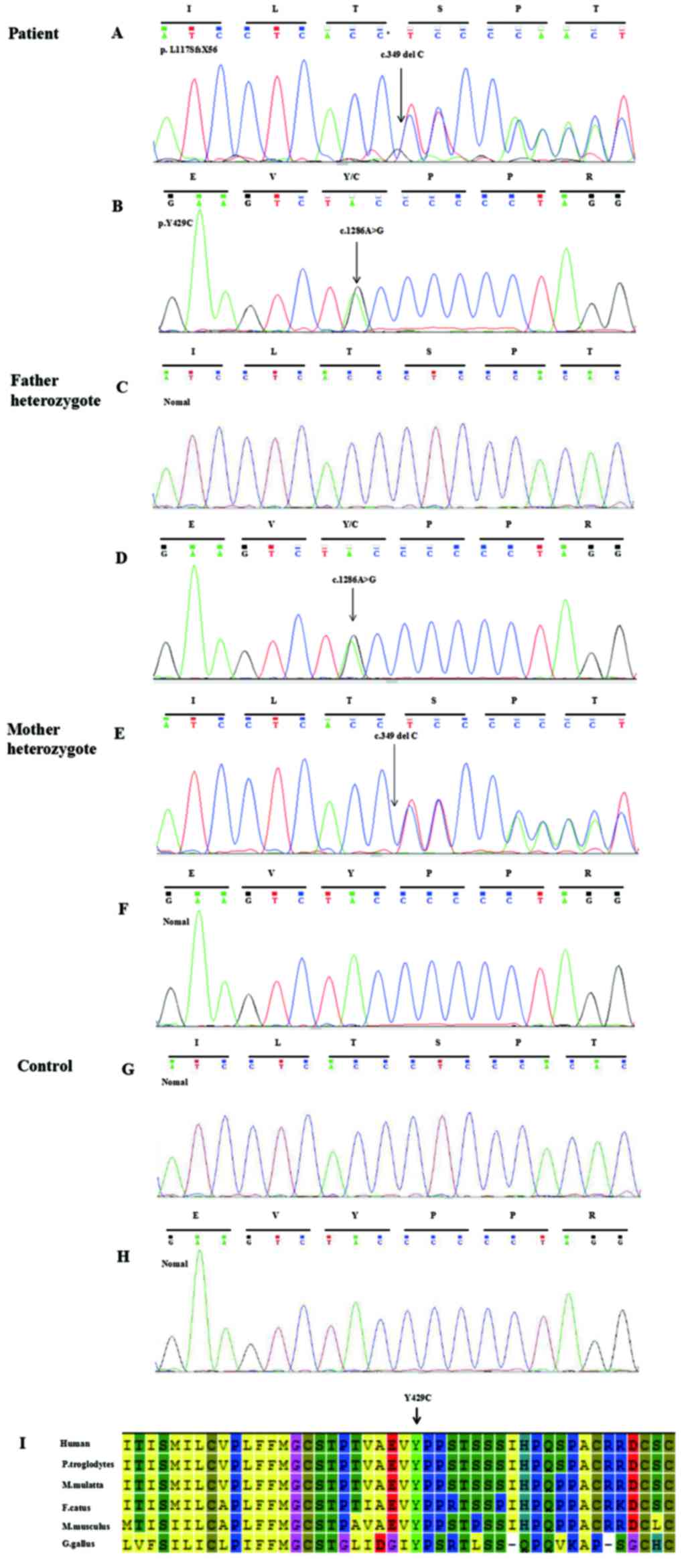 | Figure 2.Genetic analysis of the solute carrier
organic anion transporter family, member 2A1 gene in the patient,
his parents and a control. (A and B) A compound heterozygous
mutation in the patient: (A) c.349delC (p.L117SfsX56) in exon 3;
and (B) c.1286A>G (p.Y429C) in exon 9. (C and D) Heterozygote,
c.1286A>G (p.Y429C), was identified in the patient's father. (E
and F) Heterozygote, c.349delC (p.L117SfsX56), was identified in
the patient's mother. (G and H) No mutations were observed in the
control. (I) p.Y429C was revealed to be highly evolutionarily
conserved in diverse species. M. mulatta, Macaca mulatta; F.
catus, Felis catus; M. musculus, Mus musculus; G. gallus, Gallus
gallus; P. troglodytes, Pan troglodytes. |
Discussion
As is already known, the central feature in the
pathogenesis of PHO is the failure of PGE2 degradation. However, a
number of genes are directly involved in the biosynthesis and
signaling pathway of the PGE2, including HPGD,
prostaglandin-endoperoxidase synthase (PTGS) 1,
PTGS2, prostaglandin E synthase (PTGES),
PTGES2, PTGES3, PTGER1, prostaglandin E
receptor (PTGER) 2, PTGER3, PTGER4,
SLCO2A1, SLCO3A1, SLCO4A1, prostaglandin
reductase (PTGR) 1 and PTGR2 (7). Among these, Zhang et al
(6) first demonstrated in 2012
that SLCO2A1 is a PHO-pathogenic gene. The SLCO2A1
gene is located on chromosome 3q21 and organized into 14 exons,
which encode a 643 amino acid, 12-transmembrane-domain organic
anion cell-surface transporter.
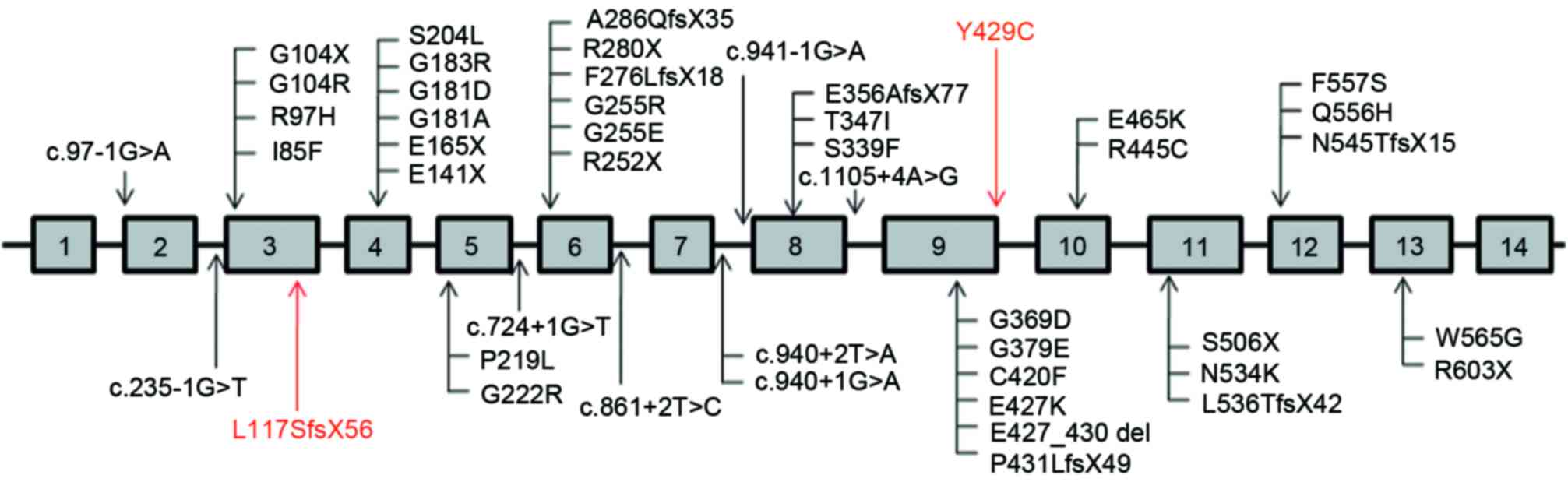
To date, 45 different SLCO2A1 mutations,
including 30 missense/nonsense mutations, eight splice sites and
seven indels, have been identified in patients with PHO. As shown
in Fig. 3, the eight splice sites
were predominantly concentrated between exons 7 and 8, and the
majority of other mutations were positioned on exons 4, 6 and 9. In
the present study, two novel mutations were identified in the
SLCO2A1 gene in PHO disease, comprising
c.349delC/p.L117SfsX56 in exon 3 and c.1286A>G/p.Y429C in exon
9. One of these mutations, c.1286A>G (p.Y429C), was identified
within the region of a previously reported deletion (11), c.1279_1290del12 (p.E427_P430del).
Sasaki et al (11) reported
that the amino acid sequence containing the p.E427_P430del mutation
is located in the extracellular region between the 9th and 10th
transmembrane domains, and this mutation may have a less severe
effect on PG transport activity (12), which may be consistent with the
faint pachydermia. The patient examined in the present study was
consistent with the report by Sasaki et al (11), describing an incomplete form of
PHO, including finger clubbing and periostosis, but lacking cutis
verticis gyrate (CVG).
With the exception of the three major phenotypes of
clubbing, periostosis and pachydermia, additional symptoms,
including sebaceous hyperplasia, hyperhidrosis, blepharoptosis and
arthropathy have also been reported (13,14).
It has also been reported that, compared with the clinical spectrum
of patients affected with HPGD mutations, the clinical
manifestations in patients with SLCO2A1 mutations emerge
later, beginning with clubbing of the distal phalanges during
puberty and pachydermia shortly following puberty. However, the
degree of arthritis, joint involvement and pachydermia in patients
affected by SLCO2A1 mutations appear to be more pronounced,
compared with that in individuals with homozygous or compound
heterozygous HPGD mutations (7). In the present study, the patient
gradually presented with joint discomfort and affected activity,
however, hyperhidrosis, sebaceous hyperplasia and blepharoptosis
were not present. Hyperhidrosis, sebaceous hyperplasia and
blepharoptosis can all fall under skin symptoms. The pathogenetic
mechanism of skin symptoms involve epidermal and sebaceous gland
hyperplasia, and dermis hypoplasia, and these cell proliferation
differences between the dermis and epidermis determine the
magnitude of the affected skin in PHO (11,15,16).
Due to the delayed clinical manifestations in the patient with
SLCO2A1 mutations in the present study, it is not possible
to exclude the possibility of slowly emerging skin symptoms
following puberty. Thus, close regular follow-up is required.
A previous study has reported that
SLCO2A1-deficient patients may present with chronic anemia
secondary to hypocellular myelofibrosis (17). The incidence of anemia in Caucasian
patients with PHO was found to be almost 50% (10/19). The possible
mechanism may be associated with complex pleiotropic effects of
PGE2 on multilineage and lineage-restricted hematopoietic
progenitor cells (18), however,
in the present study and other studies (1,6,19),
the incidence of anemia in Chinese patients is low. This data may
reflect ethnic differences.
Another specific symptom reported is
gastrointestinal discomfort. Common gastrointestinal involvement
includes chronic gastritis, hypertrophic gastropathy, peptic ulcer,
Crohn's disease and watery diarrhea (1). The patient in the present study
denied any gastrointestinal symptoms. Thus, gastroscopy, gastric
mucosa biopsy and associated gastrointestinal system examinations
were not performed. However, considering hypertrophic gastropathy,
this disease often lacks typical symptoms and the majority of
patients with hypertrophic gastropathy often show no symptoms,
despite the presence of gastric mucosal lesions and hypertrophy on
gastroscopy observation. Thus, additional specimen collection and
analyses is required to analyze the incidence and prevalence of
hypertrophic gastropathy in PHO.
In conclusion, the present study identified two
novel mutations (c.349delC/p.L117SfsX56 and c.1286A>G/p.Y429C)
in SLCO2A1 in a patient with PHO. These mutations of
SLCO2A1 (p.L117SfsX56 and p.Y429C) are the first, to the
best of our knowledge, to be reported in PHO, and the finding
provides clues to phenotype-genotype associations. In addition, the
findings obtained in the present study are consistent with those of
other studies and suggests that p.Y429C may have a less severe
effect on PG transport activity, and may be consistent with the
faint pachydermia. The results of the present study expand the
mutant spectrum of PHO, which contributes to a more rapid genetic
diagnosis, and interpretation of genetic information for prenatal
diagnosis and genetic counseling.
Acknowledgements
The authors would like to thank the State Key
Laboratory of Medical Genetics of China for their technical
assistance. This study was supported by the National Natural
Science Foundation of China (grant nos. 81470202 and 81570288), the
Natural Science Foundation of Hunan Province, China (grant nos.
13JJ3020 and 2015JC3032), the Scientific and Technological Project
of Changsha City (grant no. K1406020-31) and the National Key
Clinical Specialty Construction Projects of China.
References
|
1
|
Zhang Z, He JW, Fu WZ, Zhang CQ and Zhang
ZL: Mutations in the SLCO2A1 gene and primary hypertrophic
osteoarthropathy: A clinical and biochemical characterization. J
Clin Endocrinol Metab. 98:E923–E933. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Oikarinen A, Palatsi R, Kylmäniemi M,
Keski-Oja J, Risteli J and Kallioinen M: Pachydermoperiostosis:
Analysis of the connective tissue abnormality in one family. J Am
Acad Dermatol. 31:947–953. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rimoin DL: Pachydermoperiostosis
(idiopathic clubbing and periostosis): Genetic and physiologic
considerations. N Engl J Med. 272:923–931. 1965. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jajic I: Epidemiology of hypertrophic
osteoarthropathy. Clin Exp Rheumatol. 10:(Suppl 7). S131992.
|
|
5
|
Uppal S, Diggle CP, Carr IM, Fishwick CW,
Ahmed M, Ibrahim GH, Helliwell PS, Latos-Bieleńska A, Phillips SE,
Markham AF, et al: Mutations in 15-hydroxyprostaglandin
dehydrogenase cause primary hypertrophic osteoarthropathy. Nat
Genet. 40:789–793. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang Z, Xia W, He J, Zhang Z, Ke Y, Yue
H, Wang C, Zhang H, Gu J, Hu W, et al: Exome sequencing identifies
SLCO2A1 mutations as a cause of primary hypertrophic
osteoarthropathy. Am J Hum Genet. 90:125–132. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Seifert W, Kühnisch J, Tüysüz B, Specker
C, Brouwers A and Horn D: Mutations in the prostaglandin
transporter encoding gene SLCO2A1 cause primary hypertrophic
osteoarthropathy and isolated digital clubbing. Hum Mutat.
33:660–664. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tan ZP, Huang C, Xu ZB, Yang JF and Yang
YF: Novel ZFPM2/FOG2 variants in patients with double outlet right
ventricle. Clin Genet. 82:466–471. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sunyaev S, Ramensky V and Bork P: Towards
a structural basis of human non-synonymous single nucleotide
polymorphisms. Trends Genet. 16:198–200. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schwarz JM, Rödelsperger C, Schuelke M and
Seelow D: Mutation taster evaluates disease-causing potential of
sequence alterations. Nat Methods. 7:575–576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sasaki T, Niizeki H, Shimizu A, Shiohama
A, Hirakiyama A, Okuyama T, Seki A, Kabashima K, Otsuka A, Ishiko
A, et al: Identification of mutations in the prostaglandin
transporter gene SLCO2A1 and its phenotype-genotype correlation in
Japanese patients with pachydermoperiostosis. J Dermatol Sci.
68:36–44. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Minakawa S, Kaneko T, Niizeki H, Mizukami
H, Saito Y, Nigawara T, Kurose R, Nakabayashi K, Kabashima K and
Sawamura D: Case of pachydermoperiostosis with solute carrier
organic anion transporter family, member 2A1 (SLCO2A1) mutations. J
Dermatol. 42:908–910. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dias Madruga JA, Rosa RS, Perpétuo I,
Rodrigues AM, Janeiro A, Costa MM, Gaião L, da Pereira Silva JA,
Fonseca JE and Miltenberger-Miltenyi G: Pachydermoperiostosis in an
African patient caused by a Chinese/Japanese SLCO2A1 mutation-case
report and review of literature. Semin Arthritis Rheum. 43:566–569.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang Z, He JW, Fu WZ, Zhang CQ and Zhang
ZL: Two novel mutations in the SLCO2A1 gene in a Chinese patient
with primary hypertrophic osteoarthropathy. Gene. 534:421–423.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Neufang G, Furstenberger G, Heidt M, Marks
F and Müller-Decker K: Abnormal differentiation of epidermis in
transgenic mice constitutively expressing cyclooxygenase-2 in skin.
Proc Natl Acad Sci USA. 98:7629–7634. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Weinberg E, Topaz M, Dard M, Lyngstadaas
P, Nemcovsky C and Weinreb M: Differential effects of prostaglandin
E(2) and enamel matrix derivative on the proliferation of human
gingival and dermal fibroblasts and gingival keratinocytes. J
Periodontal Res. 45:731–740. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Diggle CP, Parry DA, Logan CV, Laissue P,
Rivera C, Restrepo CM, Fonseca DJ, Morgan JE, Allanore Y, Fontenay
M, et al: Prostaglandin transporter mutations cause
pachydermoperiostosis with myelofibrosis. Hum Mutat. 33:1175–1181.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
North TE, Goessling W, Walkley CR,
Lengerke C, Kopani KR, Lord AM, Weber GJ, Bowman TV, Jang IH,
Grosser T, et al: Prostaglandin E2 regulates vertebrate
haematopoietic stem cell homeostasis. Nature. 447:1007–1011. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Z, He JW, Fu WZ, Zhang CQ and Zhang
ZL: A novel mutation in the SLCO2A1 gene in a Chinese family with
primary hypertrophic osteoarthropathy. Gene. 521:191–194. 2013.
View Article : Google Scholar : PubMed/NCBI
|